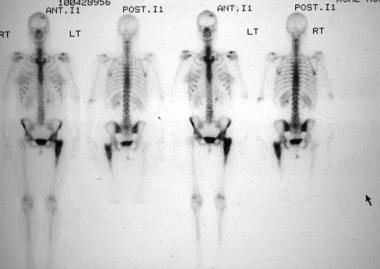
Systemic lupus erythematosus (SLE) is a chronic inflammatory disease that has protean manifestations and follows a relapsing and remitting course. More than 90% of cases of SLE occur in women, frequently starting at childbearing age. See the image below.
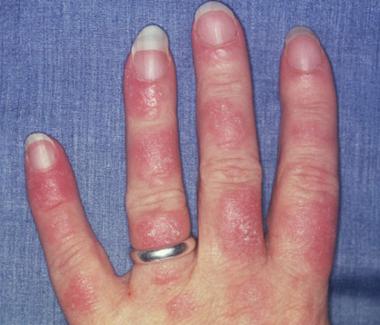 Photosensitive systemic lupus erythematosus (SLE) rashes typically occur on the face or extremities, which are sun-exposed regions. Although the interphalangeal spaces are affected, the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) and distal interphalangeal (DIP) joints are spared. Photo courtesy of Dr. Erik Stratman, Marshfield Clinic.
Photosensitive systemic lupus erythematosus (SLE) rashes typically occur on the face or extremities, which are sun-exposed regions. Although the interphalangeal spaces are affected, the metacarpophalangeal (MCP) and proximal interphalangeal (PIP) and distal interphalangeal (DIP) joints are spared. Photo courtesy of Dr. Erik Stratman, Marshfield Clinic.
See Cutaneous Clues to Accurately Diagnosing Rheumatologic Disease, a Critical Images slideshow, to help recognize cutaneous manifestations of rheumatologic diseases. Also, see the Autoimmune Disorders: Making Sense of Nonspecific Symptoms slideshow to help identify several diseases that can cause a variety of nonspecific symptoms.
SLE is a chronic autoimmune disease that can affect almost any organ system; thus, its presentation and course are highly variable, ranging from indolent to fulminant.
In childhood-onset SLE, there are several clinical symptoms more commonly found than in adults, including malar rash, ulcers/mucocutaneous involvement, renal involvement, proteinuria, urinary cellular casts, seizures, thrombocytopenia, hemolytic anemia, fever, and lymphadenopathy.[1]
In adults, Raynaud pleuritis and sicca are twice as common as in children and adolescents.[1]
The classic presentation of a triad of fever, joint pain, and rash in a woman of childbearing age should prompt investigation into the diagnosis of SLE.[2, 3]
Patients may present with any of the following manifestations[4] :
In patients with suggestive clinical findings, a family history of autoimmune disease should raise further suspicion of SLE.
See Clinical Presentation for more detail.
The diagnosis of SLE is based on a combination of clinical findings and laboratory evidence. Familiarity with the diagnostic criteria helps clinicians to recognize SLE and to subclassify this complex disease based on the pattern of target-organ manifestations.
The presence of 4 of the 11 American College of Rheumatology (ACR) criteria yields a sensitivity of 85% and a specificity of 95% for SLE.[5, 6]
When the Systemic Lupus International Collaborating Clinics (SLICC) group revised and validated the ACR SLE classification criteria in 2012, they classified a person as having SLE in the presence of biopsy-proven lupus nephritis with ANA or anti-dsDNA antibodies or if 4 of the diagnostic criteria, including at least 1 clinical and 1 immunologic criterion, have been satisfied.[7]
The following are the ACR diagnostic criteria in SLE, presented in the "SOAP BRAIN MD" mnemonic:
Testing
The following are useful standard laboratory studies when SLE is suspected:
Other laboratory tests that may be used in the diagnosis of SLE are as follows:
Imaging studies
The following imaging studies may be used to evaluate patients with suspected SLE:
Procedures
Procedures that may be performed in patients with suspected SLE include the following:
See Workup for more detail.
Management of SLE often depends on the individual patient’s disease severity and disease manifestations,[8] although hydroxychloroquine has a central role for long-term treatment in all SLE patients.
Pharmacotherapy
Medications used to treat SLE manifestations include the following:
See Treatment and Medication for more detail.
NextSystemic lupus erythematosus (SLE) is a chronic inflammatory disease that has protean manifestations and follows a relapsing and remitting course. It is characterized by an autoantibody response to nuclear and cytoplasmic antigens. SLE can affect any organ system, but it mainly involves the skin, joints, kidneys, blood cells, and nervous system (see Presentation).
The diagnosis of SLE must be based on the proper constellation of clinical findings (see the image below) and laboratory evidence. The American College of Rheumatology (ACR) criteria, proposed in 1982 and revised in 1997, summarize features that may aid in the diagnosis. (See Workup.) Management of this condition depends on the disease severity and organ involvement. Periodic follow-up and laboratory testing are imperative to detect signs and symptoms of new organ-system involvement and to monitor the response or adverse reactions to therapies. (See
Treatment.)
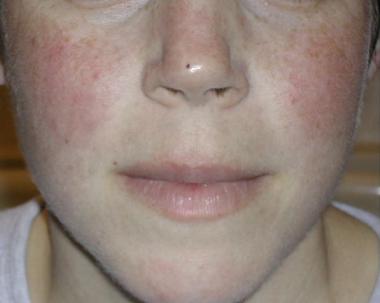 The classic malar rash, also known as a butterfly rash, with distribution over the cheeks and nasal bridge. Note that the fixed erythema, sometimes with mild induration as seen here, characteristically spares the nasolabial folds.
The classic malar rash, also known as a butterfly rash, with distribution over the cheeks and nasal bridge. Note that the fixed erythema, sometimes with mild induration as seen here, characteristically spares the nasolabial folds.
See also the following Medscape articles:
SLE is an autoimmune disorder characterized by multisystem inflammation with the generation of autoantibodies. Although the specific cause of SLE is unknown, multiple factors are associated with the development of the disease, including genetic, epigenetic, ethnic, immunoregulatory, hormonal, and environmental factors.[9, 10, 11, 12] Many immune disturbances, both innate and acquired, occur in SLE (see the image below).
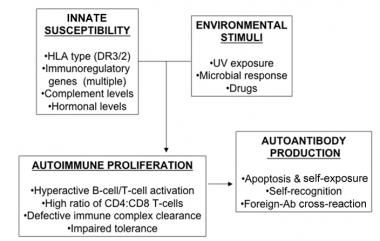 In systemic lupus erythematosus (SLE), many genetic-susceptibility factors, environmental triggers, antigen-antibody (Ab) responses, B-cell and T-cell interactions, and immune clearance processes interact to generate and perpetuate autoimmunity. HLA = human leukocyte antigen; UV = ultraviolet light.
In systemic lupus erythematosus (SLE), many genetic-susceptibility factors, environmental triggers, antigen-antibody (Ab) responses, B-cell and T-cell interactions, and immune clearance processes interact to generate and perpetuate autoimmunity. HLA = human leukocyte antigen; UV = ultraviolet light.
It is important to note that antibodies may be present for many years before the onset of the first symptoms of SLE.[13] One longstanding proposed mechanism for the development of autoantibodies involves a defect in apoptosis that causes increased cell death and a disturbance in immune tolerance.[14, 15, 10, 16] The redistribution of cellular antigens during necrosis/apoptosis leads to a cell-surface display of plasma and nuclear antigens in the form of nucleosomes. Subsequently, dysregulated (intolerant) lymphocytes begin targeting normally protected intracellular antigens. The defective clearance of the apoptotic cell debris allows for the persistence of antigen and immune complex production.[17]
T cells have long been thought to play a central role in SLE pathogenesis, and T cells from patients with lupus show defects in both signaling and effector function.[18, 19] These T cells secrete less interleukin (IL)-2, and one defect in signaling seems to be linked to an increase in calcium influx, possibly due to changes in the CD3 signaling subunits. The following seem to be adversely affected in T cells from patients with SLE: effector activity such as CD8 cytotoxicity; T-regulatory, B-cell help; migration; and adhesion. However, the method by which each of these deficits contributes to the exact clinical syndrome seen in an individual patient is still unknown. These T-cell abnormalities are currently being explored as targets for therapy, as seen with the recent approval of belimumab, which targets the B-lymphocyte stimulator (BLys) signaling pathway.[18, 19]
Many clinical manifestations of SLE are mediated by circulating immune complexes that form with antigens in various tissues or the direct effects of antibodies to cell surface components. Immune complexes form in the microvasculature, leading to complement activation and inflammation. Moreover, antibody-antigen complexes deposit on the basement membranes of skin and kidneys. In active SLE, this process has been confirmed by demonstration of complexes of nuclear antigens such as DNA, immunoglobulins, and complement proteins at these sites.
Autoantibodies have been found to be biomarkers for future neuropsychiatric events in SLE. A prospective study (=10 years) of 1047 SLE patients demonstrated that individuals who had evidence of lupus anticoagulant (LA) had an increased future risk of intracranial thrombosis and that those with anti-ribosomal P antibodies had an increased future risk of lupus psychosis.[20]
Serum antinuclear antibodies (ANAs) are found in nearly all individuals with active SLE. Antibodies to native double-stranded DNA (dsDNA) are relatively specific for the diagnosis of SLE. Whether polyclonal B-cell activation or a response to specific antigens exists is unclear, but much of the pathology involves B cells, T cells, and dendritic cells. Cytotoxic T cells and suppressor T cells (which would normally down-regulate immune responses) are decreased. The generation of polyclonal T-cell cytolytic activity is impaired. Helper (CD4+) T cells are increased. A lack of immune tolerance is observed in animal lupus models. Reports pointing to important roles of interferon-alpha, transcription factors, and signaling variations also point to a central role for neutrophils.[21]
There is a clear genetic component in SLE, with a sibling risk ratio 8-fold to 29-fold higher than that in the general population and a 10-fold increase in disease concordance in identical twins. In addition, there is a 24-56% concordance rate in monozygotic twins, compared with a 2-5% risk in dizygotic twins.[22]
Although some single genes have been implicated to play a causative role in SLE, current knowledge points toward a large number of genes being involved in a multifactorial-type inheritance pattern in most patients.[23, 24] Genome-wide association studies have identified more than 60 risk loci for SLE susceptibility across populations, with most of the genetic risk shared across borders and ethnicities.[25]
Many of the loci with a strong association with SLE are involved in the immune and related biologic systems.[22] Genes previously associated with other autoimmune diseases have been associated with SLE (eg, PTPN22 and diabetes; STAT4 and rheumatoid arthritis).
Genetic studies point to disruptions in lymphocyte signaling, interferon response, clearance of complement and immune complexes, apoptosis, and DNA methylation.[26] Several genes associated with T-cell function and signaling have been associated with SLE, including PTPN22, TNFSF4, PDCD1, IL10, BCL6, IL16, TYK2, PRL, STAT4, and RASGRP3, as have immune-complex processing and innate immunity genes, including several complement genes (eg, C2, C4A, and C4B).[27]
A meta-analysis of the association of interferon regulatory factor 5 (IRF5) with SLE found that a specific T allele, IRF5 rs2004640, is significantly associated with SLE in populations of European, Asian, and Latin American origins, whereas the A allele IRF5 rs10954213 is associated with SLE in patients of European origin but not in those of Asian origin.[28] Overall, the IRF5 gene polymorphism was found to be associated with SLE in multiple ethnic populations. The results also offer insights into the epigenetics of SLE: Hypomethylation (a form of epigenetic modification) of genes involved in osmotic lysis, apoptosis, inflammation, and cytokine pathways, among other immunologic functions, have been associated with this disease.[28]
For further discussion, see Genetics of Systemic Lupus Erythematosus
Although the specific cause of SLE is unknown, multiple genetic predispositions and gene-environment interactions have been identified (see the chart in the image below). This complex situation perhaps explains the variable clinical manifestations in persons with SLE.
In systemic lupus erythematosus (SLE), many genetic-susceptibility factors, environmental triggers, antigen-antibody (Ab) responses, B-cell and T-cell interactions, and immune clearance processes interact to generate and perpetuate autoimmunity. HLA = human leukocyte antigen; UV = ultraviolet light.
SLE has a modest recurrence rate in families: 8% of affected patients have at least one first-degree family member (parents, siblings, and children) with SLE; this is in contrast to 0.08% of the general population.[29] In addition, SLE occurs in both twins in 24% of identical twins and 2% of nonidentical twins, which may be due to a combination of genetic and environmental factors.[29]
Some studies have synthesized what is known about the mechanisms of SLE disease and genetic associations.[10, 26, 30] At least 35 genes are known to increase the risk of SLE.[26] A genetic predisposition is supported by 40% concordance in monozygotic twins; if a mother has SLE, her daughter's risk of developing the disease has been estimated to be 1:40, and her son's risk, 1:250.[26, 30]
A genome-wide study in a northern European population replicated the association of SLE with susceptibility genes related to B-cell receptor pathway signaling, as well as confirmed the association of SLE with genes at the interferon regulatory factor 5 (IRF5)-TNPO3 locus.[31] The investigators also confirmed other loci associations with SLE (TNFAIP3, FAM167A-BLK, BANK1 and KIAA1542); however, it was determined that these loci had a lower significance level and a lower contribution to individual risk for SLE.[31]
Studies of human leukocyte antigens (HLAs) reveal that HLA-A1, HLA-B8, and HLA-DR3 are more common in persons with SLE than in the general population. The presence of the null complement alleles and congenital deficiencies of complement (especially C4, C2, and other early components) are also associated with an increased risk of SLE.
Numerous studies have investigated the role of infectious etiologies that may also perpetuate autoimmunity.[32] Patients with SLE have higher titers of antibodies to Epstein-Barr virus (EBV), have increased circulating EBV viral loads, and make antibodies to retroviruses, including antibodies to protein regions homologous to nuclear antigens. In patients with SLE and EBV infection, the B cells are not primarily defective; rather, the SLE/EBV phenomenon is due to a T-cell abnormality, which causes failure in normal immunoregulation of the B-cell response.[33] Viruses may stimulate specific cells in the immune network. Chronic infections may induce anti-DNA antibodies or even lupuslike symptoms, and acute lupus flares often follow bacterial infections.
Environmental and exposure-related causes of SLE are less clear. Possible early-life risk factors include the following[34] :
Other potential factors include the following:
Pregnancy can be a time when lupus initially presents or flares, although more recent data suggests that pregnancy outcomes are favorable and flares are infrequent among patients with inactive or stable mild-moderated SLE.[36]
Vitamin D is involved in both in both innate and acquired immunity, and vitamin D deficiency has been implicated in autoimmunity and the development of rheumatic diseases, including SLE.[37, 38] Young et al studied 436 individuals who reported having a relative with SLE but who did not have SLE themselves, and found that the combination of vitamin D deficiency and carriage of specific single-nucleotide polymorphisms was associated with significantly increased risk of transitioning to SLE.[39] Hu et al reported that in an Asian population, carriage of certain polymorphisms in the vitamin D receptor gene BsmI (Bb + BB genotype and B allele) can significantly increase risk for developing SLE.[40]
Estimates of the annual incidence of SLE from the 1970s to 2000s have ranged from approximately 1 to 10 per 100,000 population, while the prevalence of SLE has been estimated to range from approximately 5.8 to 130 per 100,000 population.[41]
The Lupus Foundation of America estimates prevalence to be at least 1.5 million cases,[42] which likely reflects inclusion of milder forms of the disease. A 2008 report from the National Arthritis Data Working Group estimated a prevalence of 161,000 cases of definite SLE and 322,000 cases of definite or probable SLE.[43]
The frequency of SLE varies by race and ethnicity, with higher rates reported in blacks and Hispanics. A study in the predominantly white population of Olmsted County, Minnesota found an age-adjusted prevalence of 30.5 per 100,000 population.[41] In a study of a racially diverse population in Michigan, the prevalence of SLE was 2.3-fold higher in blacks than in whites; in that study, the age-adjusted prevalence of lupus in blacks was 105.8 or 103 per 100,000 population, depending on whether the ACR or a rheumatologiist's definition of SLE was used.[44]
Registries have been established in San Francisco and New York City to provide annual prevalence estimates for Hispanics and Asians,[45] but a 2001 study found a prevalence of 100 per 100,000 Hispanics in Nogales, Arizona.[46]
In the Michigan study, the prevalence of SLE was 10-fold higher in females than in males and was over twice as high in black females as in white females, reaching 1 in 537 among black females.[44] SLE is also more frequent in Asian women than in white women.[47]
Worldwide, the prevalence of SLE varies. The highest rates of prevalence have been reported in Italy, Spain, Martinique, and the United Kingdom Afro-Caribbean population.[48] Although the prevalence of SLE is high in black persons in the United Kingdom, the disease is rarely reported in blacks in Africa, suggesting that there may be an environmental trigger, as well as a genetic basis, for disease in the UK population.[49]
Worldwide, the prevalence of SLE appears to vary by race. However, there are different prevalence rates for people of the same race in different areas of the world. The contrast between low reported rates of SLE in black women in Africa and high rates in black women in the United Kingdom suggests that there are environmental influences.[49] In general, black women have a higher rate of SLE than women of any other race, followed by Asian women and then white women.[48]
In the United States, black women are two to four times more likely to have SLE than white women.[44, 48] A review of SLE across Asia-Pacific countries revealed considerable variation in prevalence and survival rates.[50] For example, overall prevalence rates ranged from 4.3 to 45.3 per 100,000, and the overall incidence ranged from 0.9 to 3.1 per 100,000 per year. Moreover, Asians with SLE had higher rates of renal involvement than whites did, and cardiovascular involvement was a leading cause of death in Asians.[50]
More than 90% of cases of SLE occur in women, frequently starting at childbearing age.[32, 51] The use of exogenous hormones has been associated with lupus onset and flares, suggesting a role for hormonal factors in the pathogenesis of the disease.[52] The risk of SLE development in men is similar to that in prepubertal or postmenopausal women. Interestingly, in men, SLE is more common in those with Klinefelter syndrome (ie, genotype XXY), further supporting a hormonal hypothesis. In fact, a study by Dillon et al found that men with Klinefelter syndrome had a more severe course of SLE than women but a less severe course than other men.[53]
The female-to-male ratio peaks at 11:1 during the childbearing years.[54] A correlation between age and incidence of SLE mirrors peak years of female sex hormone production. Onset of SLE is usually after puberty, typically in the 20s and 30s, with 20% of all cases diagnosed during the first 2 decades of life.[55]
A review of the worldwide literature (predominantly North America, Europe, and Asia) found that the incidence of pediatric-onset SLE ranged from 0.36 to 2.5 per 100,000 per year and the prevalence ranged from 1.89 to 25.7 per 100,000.[56]
The prevalence of SLE is highest in women aged 14 to 64 years. SLE does not have an age predilection in males, although it should be noted that in older adults, the female-to-male ratio falls.[57] This effect is likely due to loss of the estrogen effect in older women.
Systemic lupus erythematosus (SLE) carries a highly variable prognosis for individual patients. The natural history of SLE ranges from relatively benign disease to rapidly progressive and even fatal disease. SLE often waxes and wanes in affected individuals throughout life, and features of the disease vary greatly between individuals.
The disease course is milder and survival rate higher in persons with isolated skin and musculoskeletal involvement than in those with renal disease[58] and CNS disease.[59] A consortium report of 298 SLE patients followed for 5.5 years noted falls in SLE Disease Activity Index 2000 (SLEDAI-2K) scores after the first year of clinical follow-up and gradual increases in cumulative mean Systemic Lupus International Collaborating Clinics (SLICC) damage index scores.[60]
It is important to distinguish between the disease activity and the damage index (irreversible organ dysfunction). Although the most effective instrument to measure SLE disease activity is still open to debate, there are several validated measures, including the Systemic Lupus Activity Measure (SLAM), SLEDAI, Lupus Activity Index (LAI), European Consensus Lupus Activity Measurement (ECLAM), and British Isles Lupus Activity Group (BILAG) Index.
Prognostic factors from the 2008 European League Against Rheumatism (EULAR) recommendations included the following[61] :
Mortality in patients with SLE has decreased over the past few decades.[62] Prior to 1955, the 5-year survival rate in SLE was less than 50%; currently, the average 10-year survival rate exceeds 90%,[63, 59] and the 15-year survival rate is approximately 80%.[64] Previously, mortality was due to the disease itself; currently, mortality is often a result of medication side effects (eg, fatal infections in individuals receiving potent immunosuppressive medications) or cardiovascular events.
Ten-year survival rates in Asia and Africa are significantly lower than those in the United States, ranging from 60-70%,[65, 66] but this may reflect detection bias of severe cases only.
Decreased mortality rates associated with SLE can be attributed to earlier diagnosis (including milder cases), improvement in disease-specific treatments, and advances in general medical care. According to the Centers for Disease Control and Prevention (CDC), however, 35% of SLE-related deaths in the United States occur in patients younger than 45 years, making this a serious issue despite declining overall mortality rates.[45]
The EULAR task force also identified the following comorbidities as increasing the risk of morbidity and mortality in patients with SLE[61] :
In 1976, Urowitz first reported bimodal mortality in early versus late SLE, noting that SLE-related deaths usually occurred within the first 5-10 years of symptom onset.[67] Mortality in the first few years of illness is typically from severe SLE disease (eg, CNS, renal, or cardiovascular involvement) or infection related to immunosuppressive treatment. Infections account for 29% of all deaths in these patients.[68]
Late deaths (after age 35 years) are generally from myocardial infarction or stroke secondary to accelerated atherosclerosis[62, 69, 63, 70] ; inflammation is central to SLE pathogenesis and plays a major role in the development and accelerated progression of atherosclerosis. Manzi et al reported that women aged 35-44 years with SLE were 50 times more likely to develop myocardial ischemia than healthy Framingham study control women.[69] The presence of lupus nephritis may increase these risks.[71] The presence of traditional and nontraditional risk factors increases the risk of cardiovascular (CVD) disease in patients with SLE.
In a study by Petri et al that evaluated a large sample of SLE patients, the investigators reported that more than 50% of these patients had at least 3 classic cardiac risk factors, with the most common ones being a sedentary lifestyle, obesity, and hypercholesterolemia.[72] In another study, Salmon et al found that nontraditional CVD risk factors in SLE patients included having higher homocysteine levels, renal impairment, enhanced LDL oxidation, and chronic inflammation.[73]
Causes of accelerated coronary artery disease in persons with SLE are likely multifactorial. They include endothelial dysfunction, inflammatory mediators, corticosteroid-induced atherogenesis, and dyslipidemia.
The influence of race on prognosis has been widely debated. The LUMINA study group examined SLE in black, white, and Hispanic patients in the United States (including Puerto Rico) and reported that both disease activity and poverty predicted higher mortality in racial and ethnic minorities.[74] In the Michigan Lupus Epidemiology and Surveillance program, the proportion of patients with renal disease was 2.2-fold higher, and that of progression to end-stage renal disease was 3.4-fold higher, in blacks than in whites.[44]
Stress the importance of adherence to medications and follow-up appointments for detection and control of SLE disease. Instruct patients with SLE to seek medical care for evaluation of new symptoms, including fever. Advise them regarding their heightened risks for infection and cardiovascular disease. Educate patients with SLE regarding aggressive lipid and blood pressure goals to minimize the risk of coronary artery disease.
Instruct patients with SLE to avoid exposure to sunlight and ultraviolet light. Also, encourage them to receive nonlive vaccines during stable periods of disease, to quit smoking, and to carefully plan pregnancies.
For patient education information, see the Arthritis Center, as well as Lupus (Systemic Lupus Erythematosus ).
See also the American College of Rheumatology’s patient fact sheets for SLE, Systemic Lupus Erythematosus in Children and Teens, and Antiphospholipid Syndrome.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved