Polymyositis is an idiopathic inflammatory myopathy that causes symmetrical, proximal muscle weakness; elevated skeletal muscle enzyme levels; and characteristic electromyography (EMG) and muscle biopsy findings (see the images below). Clinically similar to polymyositis, dermatomyositis is an idiopathic, inflammatory myopathy associated with characteristic dermatologic manifestations.[1] Inclusion body myositis is a slowly progressive, idiopathic, inflammatory myopathy with characteristic pathologic findings that is generally found in older males. Bohan and Peter classify the idiopathic inflammatory myopathies as follows[2] :
See Etiology, Presentation, and Workup.
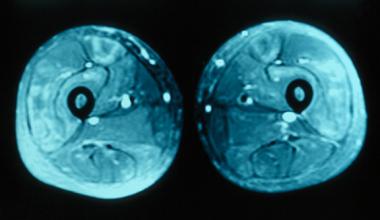 MRI of thighs showing increased signal in the quadriceps muscles bilaterally consistent with inflammatory myositis.
MRI of thighs showing increased signal in the quadriceps muscles bilaterally consistent with inflammatory myositis.
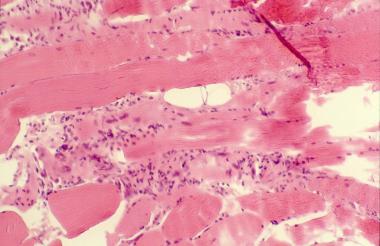 Histopathology of polymyositis showing endomysial mononuclear inflammatory infiltrate and muscle fiber necrosis.
Histopathology of polymyositis showing endomysial mononuclear inflammatory infiltrate and muscle fiber necrosis.
Polymyositis and dermatomyositis have many shared clinical features. Both are inflammatory myopathies that present as symmetrical muscle weakness that develops over weeks to months. Initial treatment with corticosteroids usually produces a response; however, nonresponders require further treatment. Moreover, both conditions may be associated with malignancies.[3] Despite these similarities, muscle biopsy findings and characteristic skin findings of dermatomyositis reveal each as a distinct clinical entity. (See Presentation, Workup, Treatment, and Medication.)
Although classified as an inflammatory myopathy, inclusion body myositis shows minimal evidence of inflammation. This is the most common inflammatory myopathy in patients older than age 50 years. It more commonly presents with asymmetrical, distal weakness and also has distinct biopsy findings. Studies so far have yielded less favorable results than treatment for polymyositis and dermatomyositis. (See Treatment and Medication.)
Patients with polymyositis should be educated early about the disease and should be provided with realistic expectations about outcomes. Most patients show significant improvement with treatment.
Stress the need for close follow-up care, continued physical therapy, and long-term therapy, and warn patients regarding adverse events related to medications. Patients may visit The Myositis Association Web site for more information.
NextPolymyositis is an immune-mediated syndrome secondary to defective cellular immunity that is most commonly associated with other systemic autoimmune diseases. It may be due to diverse causes that occur alone or in association with viral infections, malignancies, or connective-tissue disorders. Evidence points toward a T-cell–mediated cytotoxic process directed against unidentified muscle antigens. Supporting this conclusion are CD8 T cells, which, along with macrophages, initially surround healthy nonnecrotic muscle fibers and eventually invade and destroy them. (See the image below.)
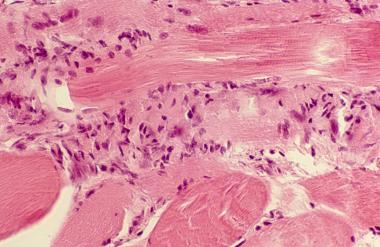 Close view of muscle biopsy, showing chronic inflammatory infiltrate consisting of T lymphocytes, especially CD8+ T lymphocytes.
Close view of muscle biopsy, showing chronic inflammatory infiltrate consisting of T lymphocytes, especially CD8+ T lymphocytes.
The factors triggering a T-cell–mediated process in polymyositis are unclear. Viruses have been implicated; so far, however, only the human retroviruses human immunodeficiency virus (HIV) and human T-cell lymphotrophic virus type I (HTLV-I), the simian retroviruses, and coxsackievirus B have been etiologically connected with the disease. These viruses may directly invade the muscle tissue, damaging the vascular endothelium and releasing cytokines, which then induce abnormal expression of the major histocompatibility complex (MHC) and render the muscle susceptible to destruction.
An autoimmune response to nuclear and cytoplasmic autoantigens is detected in about 60-80% of patients with polymyositis and dermatomyositis. Some serum autoantibodies are shared with other autoimmune diseases (ie, myositis-associated antibodies [MAAs]), and some are unique to myositis (ie, myositis-specific antibodies [MSAs]). The MSAs are found in approximately 40% of patients with polymyositis or dermatomyositis, whereas MAAs are found in 20-50% of these patients.
The identified MSA targets include 3 distinct groups of proteins: aminoacyl–transfer ribonucleic acid (tRNA) synthetases (anti-Jo-1), nuclear Mi-2 protein, and components of the signal-recognition particle (SRP).
Most of the anti-tRNA synthetase antibodies are directed toward functional and highly conserved domains of the enzyme. As many as 6 of 20 aminoacyl-tRNA synthetases have been described, but anti-histidyl-tRNA synthetase (Jo-1) is most common (20-30%). Autoantibodies directed toward the other synthetases specific for alanine (anti-PL12), glycine (anti-EJ), isoleucine (anti-OJ), threonine (anti-PL7), and asparagine (anti-KS) have been reported in only about 1% of patients.
Anti-Jo-1 autoantibodies were originally described as precipitating autoantibodies in sera of patients with polymyositis. Later, the anti-Jo-1 antibodies were recognized to be specific for patients with polymyositis. The target for the anti-Jo-1 antibodies was the aminoacyl-tRNA synthetases, a family of distinct cellular enzymes.
The Jo-1 antigen is histidyl-tRNA synthetase. This enzyme is partially responsible for attaching tRNA to their cognate ribosomal RNA (rRNA). The Jo-1 antigen migrates as a 53-kd protein on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
The presence of autoantibodies against the Jo-1 antigen has been reported in up to 23% of polymyositis patients by immunodiffusion. Anti–Jo-1 antibodies are almost completely specific for myositis and are more common in polymyositis than in dermatomyositis; they are rare in children. The presence of anti-Jo-1 antibodies defines a distinct group of polymyositis patients with interstitial lung disease, arthritis, and fevers. The anti–Jo-1 response appears to be self-antigen driven, having a broad spectrotype over time and undergoing isotype switching. Anti–Jo-1 antibodies also inhibit the function of histidyl-tRNA synthetase in humans more than they do in other species.
Anti-Mi-2 antibodies recognize a major protein of a nuclear complex formed by at least 7 proteins that is involved in the transcription process. Autoantibodies recognizing Mi-2 are considered specific serologic markers of dermatomyositis. They are detected in about 20% of patients with myositis and are associated with relatively acute onset, a good prognosis, and a good response to therapy.
Anti-SRP antibodies are directed toward an RNA-protein complex that consists of 6 proteins and a 300-nucleotide RNA molecule (7SL RNA). Patients with anti-SRP antibodies have acute polymyositis with cardiac involvement, a poor prognosis, and a poor response to therapy.
The MAA are found in the sera of 20-50% of patients and are commonly encountered in other connective tissue diseases. The most important antigenic targets of the MAA are the PM/Scl nucleolar antigen, the nuclear Ku antigen, the small nuclear ribonucleoproteins (snRNP), and the cytoplasmic ribonucleoproteins (RoRNP). The anti-PM/Scl autoantibodies are generally found in patients affected by polymyositis overlapping with scleroderma. Anti-Ku antibodies are found in patients with myositis overlapping with other connective tissue diseases.
Antibodies directed against snRNP are frequently found in patients with myositis and in patients with connective tissue–disease overlap syndrome, whereas antibodies toward Ro/SSA 60 kD, Ro/SSA 52 kD, and La/SSB protein components of the RoRNP complex are almost exclusively found in patients with Sjögren syndrome and systemic lupus erythematosus (SLE).
An increased association of myositis has been found with human leukocyte antigen (HLA) haplotypes A1, B8, and DR3, which also increase the risk for autoimmune diseases. Environmental triggers, especially infectious agents, have been suggested as etiologic agents. These include the following:
Many drugs are known to cause myopathy. Most drugs, such as hydroxychloroquine and colchicine, cause a toxic or metabolic myopathy.
Several drugs, however, rarely induce an immune-mediated myopathy or myositis. Muscle biopsy shows chronic inflammatory changes consistent with polymyositis. Drugs such as D-penicillamine, hydralazine, procainamide, phenytoin, and angiotensin-converting enzyme (ACE) inhibitors have been associated with this type of inflammatory myopathy. Statins occasionally cause severe muscle inflammation and rhabdomyolysis.
Idiopathic inflammatory myopathies are relatively rare diseases, with an incidence in the United States that ranges from 0.5-8.4 cases per million population. Polymyositis is more common in the United States within the black population, with the estimated black-to-white incidences for polymyositis and dermatomyositis being 5:1 and 3:1, respectively. Internationally, polymyositis is less common among Japanese persons.
Polymyositis and dermatomyositis are more common in women than in men (2:1 ratio), while inclusion body myositis is twice as common in men.
Polymyositis usually affects adults older than 20 years, especially those aged 45-60 years. Polymyositis rarely affects children. The age of onset of polymyositis with another collagen vascular disease is related to the associated condition.
Although dermatomyositis is primarily a disease of adults, it also is observed in children, usually those aged 5-14 years. Eighty percent of patients with inclusion body myositis are older than 50 years at onset.
In most patients, polymyositis responds well to treatment, although residual weakness occurs in approximately 30% of patients. Osteoporosis, a common complication of long-term corticosteroid therapy, may cause significant morbidity. A study from Taiwan determined that the risk of osteoporosis was 2.99 times higher in patients with polymyositis, and that the risk was independent of corticosteroid and immunosuppressant treatment.[4]
Poor prognostic factors include the following:
Complications of polymyositis may include the following:
Carruthers et al reported that patients with polymyositis are at increased risk for venous thromboembolism (VTE), with hazard ratios of 7.0 for VTE, 6.16 for deep venous thrombosis, and 7.23 for pulmonary embolism. Overall, the highest calculated incidence rate ratios were observed in the first year after diagnosis of polymyositis.[8]
The incidence of lung, bladder, and non-Hodgkin lymphoma may be increased in patients with polymyositis, especially in the first year after diagnosis.
Five-year survival rates have been estimated at more than 80%. Mortality is most often related to associated malignancy or pulmonary complications; however, elderly patients with cardiac involvement or dysphagia also have a higher mortality rate.[9]
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved