Limb-girdle muscular dystrophy refers to a group of disorders that cause weakness and wasting of the muscles around the shoulders and hips.
All patients have a history of progressive, symmetric proximal muscle weakness that usually starts in childhood to young adulthood. Pelvic muscle weakness is most often the first symptom. Other symptoms may include the following:
See Clinical Presentation for more detail.
Muscle biopsy and genetic testing are the most important tools used in the diagnostic evaluation of patients in whom limb-girdle muscular dystrophy (LGMD) is suspected.
Creatine kinase is also used for diagnosis because autosomal recessive LGMDs often cause extremely high CK levels.
Magnetic resonance imaging (MRI) can help differentiate forms of LGMD.
See Workup for more detail.
No specific treatment is available for any of the LGMD syndromes, though aggressive supportive care is essential to preserve muscle function, maximize functional ability, and prolong life expectancy.
Aggressive use of passive stretching, bracing, and orthopedic procedures allow the patient to remain independent for as long as possible.
Orthopedic surgery may be needed to help correct or prevent contractures and scoliosis.
See Treatment and Medication for more detail.
NextWalton and Nattrass first proposed limb-girdle muscular dystrophy (LGMD) as a nosological entity in 1954.[1] Their definition included the following characteristics:
This classification allowed for a wide range of phenotypic variability. Improved diagnostic methods have demonstrated that a large group of neuromuscular disorders, including some that were not truly LGMD, were included in this definition.
LGMD classification has been revolutionized with the advent of molecular genetics. The most common classification scheme is based on clinical and molecular characteristics. It divides cases into autosomal dominant (LGMD1) and autosomal recessive (LGMD2) syndromes. This list continues to expand, and, as of this writing, specific mutations listed on OMIM are known for 7 autosomal dominant LGMDs and 23 autosomal recessive LGMDs. Overlap exists with Congenital Muscular Dystrophy (CMD) as several gene mutations can cause both a LGMD and CMD phenotype.
Although not truly limb-girdle syndromes, diseases classified as myofibrillar myopathies share several phenotypic characteristics with the LGMDs. They are usually adult-onset diseases with slowly progressive weakness involving proximal (and distal) muscles. Many patients have respiratory failure, cardiomyopathy, and neuropathy. Some mutations can cause both a myofibrillar myopathy and a muscular dystrophy phenotype.
Cardinal morphologic features of myofibrillar myopathies on muscle biopsy are vacuolated muscle fibers and inclusions that were initially given different names in the 1970s. In 1980, desmin was noted to accumulate in the inclusions, and the name desmin storage myopathy was coined. However, in the mid 1990s, other proteins were also found to accumulate in the abnormal muscle fibers, and molecular genetic studies revealed several chromosomal loci. Since then, the relatively generic term myofibrillar myopathy has been adopted. These diseases are discussed here in part because mutations in 2 genes can present with either an LGMD or a myofibrillar myopathy phenotype.
Limb-girdle muscular dystrophy (LGMD) protein defects occur in several pathways involved in the biologic function of muscle and can be divided into groups based on cellular localization. These include proteins associated with the sarcolemma (see image below), proteins associated with the contractile apparatus (see image below), and various enzymes involved in muscle function. However, although the primary defect in many LGMDs is known, the precise mechanism leading to the dystrophic phenotype has not always been elucidated. Specific protein function and abnormalities are discussed below with each LGMD.
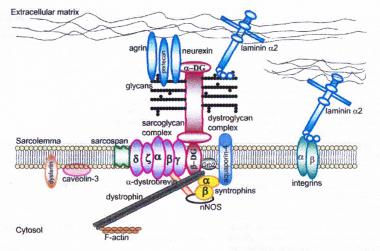 Dystrophin-glycoprotein complex bridges the inner cytoskeleton (F-actin) and the basal lamina. Mutations in all sarcoglycans, dysferlin, and caveolin-3, as well as mutations that cause abnormal glycosylation of alpha-dystroglycan can result in limb-girdle muscular dystrophy syndrome. Reprinted with permission from Cohn RD. Dystroglycan: important player in skeletal muscle and beyond. In: Neuromuscular Disorders. Vol. 15. Cohn RD. Elsevier; 2005: 207-17. 7, 20
Dystrophin-glycoprotein complex bridges the inner cytoskeleton (F-actin) and the basal lamina. Mutations in all sarcoglycans, dysferlin, and caveolin-3, as well as mutations that cause abnormal glycosylation of alpha-dystroglycan can result in limb-girdle muscular dystrophy syndrome. Reprinted with permission from Cohn RD. Dystroglycan: important player in skeletal muscle and beyond. In: Neuromuscular Disorders. Vol. 15. Cohn RD. Elsevier; 2005: 207-17. 7, 20 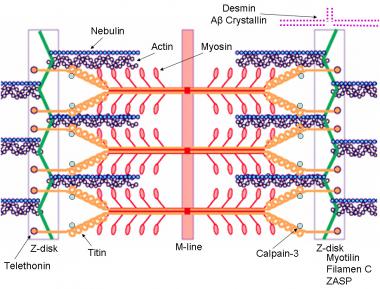 Schematic of the sarcomere with labeled molecular components that are known to cause limb-girdle muscular dystrophy or myofibrillar myopathy. Mutations in actin and nebulin cause the congenital myopathy nemaline rod myopathy, and the mutations in myosin cause familial hypertrophic cardiomyopathy. Image courtesy of Dr F. Schoeni-Affoher, University of Friberg, Switzerland.
Schematic of the sarcomere with labeled molecular components that are known to cause limb-girdle muscular dystrophy or myofibrillar myopathy. Mutations in actin and nebulin cause the congenital myopathy nemaline rod myopathy, and the mutations in myosin cause familial hypertrophic cardiomyopathy. Image courtesy of Dr F. Schoeni-Affoher, University of Friberg, Switzerland. The cause of myofibrillar myopathies is unknown but is discussed below for each of the different mutations that have been associated with the disease. Of interest, several mutations that result in myofibrillar myopathy are in genes that code for Z-disk proteins.
LGMD subtypes based on pathophysiological mechanism[2]
Autosomal recessive LGMDs are more common than the autosomal dominant forms of the disease, which probably account for about 10% of all LGMDs. The overall frequency of all LGMD syndromes has been estimated to be 5-70 per 1 million population in several countries.
Different populations often have different frequencies of the various LGMDs.
Several studies throughout the world have estimated the frequency of LGMDs based on immunochemical and genetic testing.[3, 4, 5, 6, 7] In many studies, LGMD2A is the most common, accounting for 8-26% of all LGMDs. In some populations, it may be the only LGMD present (Reunion Island, Basque Country) with very high prevalence rates (48-69 cases per million). LGMD2B is also relatively common, accounting for 3-19% of all LGMDs. LGMD2I is common in certain parts of Northern Europe (Denmark and parts of England), but worldwide frequencies outside this area account for 3-8% of all LGMDs.
The sarcoglycanopathies as a group (LGMD2C-LGMD2F) are a common cause of LGMDs, accounting for 3-18%, with a high percentage of severe cases. As with other LGMDs, different sarcoglycanopathies are overrepresented or underrepresented in different populations, with some populations having representative cases of all 4 sarcoglycanopathies and other populations having only 1 mutation type, which is probably related to founder effects and population inbreeding (consanguinity). LGMD2C is common in Tunisia; LGMD2D is common in Europe, the United States, and Brazil; and LGMD2E and LGMD2F are common in Brazil. Overall, LGMD2D (α-sarcoglycanopathy) is twice as common as LGMD2C (γ-sarcoglycanopathy) and LGMD2E (β-sarcoglycanopathy), and LGMD2F (δ-sarcoglycanopathy) is the rarest.
All the congenital muscular dystrophies can present with a LGMD phenotype, and OMIM recognizes 4 at this time (LGMD2I, LGMD2K, LGMD2M, LGMD2N).
Morbidity and mortality rates vary. With early onset, the course is generally rapid.
Patients can become wheelchair bound in their early teens and die from respiratory complications in their late teens.
Patients with slowly progressive LGMD may be able to ambulate for more than 30 years after disease onset, and they may not become wheelchair bound until much later than their teens. Some patients with confirmed mutations have had nearly normal strength.
LGMD is reported in races and countries throughout the world.
Autosomal dominant and autosomal recessive forms of LGMD affect both sexes equally.
The age of onset varies among the different mutations. It also can vary among families with the same mutation. Reported age of onset of LGMDs is between 1 and 50 years, although some patients may be asymptomatic. Myofibrillar myopathies can present in the first decade of life up until the 60s or 70s.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved