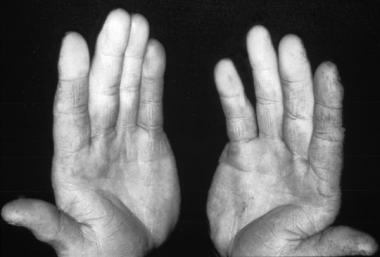
Hypertrophic osteoarthropathy (HOA) is a syndrome of clubbing of the digits, periostitis of the long (tubular) bones, and arthritis.[1] This clinical triad of digital clubbing, arthralgias, and ossifying periostitis has been recognized since the late 1800s and was previously known as hypertrophic pulmonary osteoarthropathy (HPOA). It is a syndrome characterized by excessive proliferation of skin and bone at the distal parts of extremities and by digital clubbing and periostosis of the tubular bones.[2] Hippocrates first described digital clubbing 2500 years ago, hence the use of the term Hippocratic fingers.[3] Observations made in modern times by Bamberger (1889), Pierre Marie (1890), and other investigators led to identification of various causes of this digital anomaly, which can be the first manifestation of a severe organic disease such as chronic pulmonary and cardiac diseases,[4] hence also named as Pierre Marie–Bamberger disease.[5] .
The disease is classified either as primary (hereditary or idiopathic) or secondary. Primary hypertrophic osteoarthropathy (also termed primary pachydermoperiostosis or Touraine-Solente-Gole syndrome) was initially described by Friedreich in 1868 and then by Touraine et al in 1935, who recognized its familiar features. The 3 recognized forms of primary osteoarthropathy are (1) complete (pachydermia, digital clubbing, and periostosis), (2) incomplete (no pachydermia), and (3) fruste form (prominent pachydermia with few skeletal manifestations). This classification was proposed by Touraine et al.[6, 7] Primary hypertrophic osteoarthropathy represents 3% of all cases of hypertrophic osteoarthropathy. Its prevalence in the general population is not exactly known.
Interestingly, some patients with primary hypertrophic osteoarthropathy eventually develop diseases (eg, patent ductus arteriosus, Crohn disease, myelofibrosis) that are otherwise known to be underlying causes of secondary hypertrophic osteoarthropathy, as late as 6-20 years after the onset of the osteoarthropathy.[8, 9]
Secondary hypertrophic osteoarthropathy is associated with an underlying pulmonary, cardiac, hepatic, or intestinal disease and often has a more rapid course.
Clubbing is elevation of the nail and widening of the distal phalanx caused by swelling of the subungual capillary bed resulting from increased collagen deposition, interstitial inflammation with edema, and proliferation of the capillaries themselves. Increased vascular supply to the nail bed and increased connective tissue growth, together producing the characteristic clubbing.[10] Perivascular infiltrates of lymphocytes and vascular hyperplasia are responsible for thickening of the vessel walls. Electron microscopy reveals Weibel-Palade bodies and prominent Golgi complexes, confirming structural vessel wall damage.[11] Vast numbers of arteriovenous anastomoses may also be seen in the nail bed.[12]
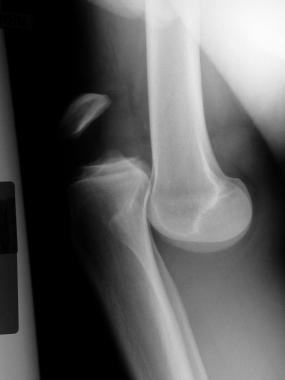
Subperiosteal new bone formation exists along the distal diaphysis of tubular bones, progressing proximally over time. The irregular periosteal proliferation affects predominantly the distal ends of long bones, including the epiphysis in 80-97% of patients. Usually the metacarpus, metatarsus, tibia, fibula, radius, ulna, femur, humerus, and clavicle are involved. The tibia is almost invariably involved.[13, 14, 15] Involvement of the epiphysis distinguishes it from the secondary form that typically spares the epiphysis.
Initially, excessive connective tissue and subperiosteal edema elevate the periosteum; then, new osteoid matrix is deposited beneath the periosteum.[13] As this mineralizes, a new layer of bone is formed, and, eventually, the distal long bones may become sheathed with a cuff of new bone.[16]
The pathological hallmark of hypertrophic osteoarthropathy is neoangiogenesis and edema and osteoblast proliferation in distal tubular bones that leads to subperiosteal new-bone formation.
Two types of bone changes can be found in the distal phalanges, hypertrophic and osteolytic.[17] Hypertrophy or bony overgrowth predominates in patients with lung cancer and HPOA, whereas acroosteolysis predominates in patients with cyanotic congenital heart disease and hypertrophic osteoarthropathy.[18] The type of bone remodeling process depends on the age when clubbing develops.[17] If clubbing appears in childhood, osteolysis is more prominent; however, if it develops after puberty, hypertrophic changes take place. Pineda et al hypothesize that a putative circulating growth factor destroys immature bone.[17]
Synovial involvement may occur with subperiosteal changes.[13] Thickening of the subsynovial blood vessels and mild lining-layer hyperplasia may occur.[19, 13] The edematous synovium becomes mildly infiltrated with lymphocytes, plasma cells, and occasional polymorphonuclear leukocytes, but the results from immunohistologic studies are negative. Electron-dense subendothelial deposits are present in vessel walls.[20, 21, 22] In a study of a patient with primary hypertrophic osteoarthropathy and chronic arthritis, Lauter et al found multilayered basement laminae around small subsynovial blood vessels consistent with the late stages of vascular injury.[22] Synovial fluid is usually noninflammatory with low leukocyte counts and few neutrophils.[20, 22]
Skin changes are more evident in primary hypertrophic osteoarthropathy and are characterized by thick skin or pachyderma which is caused by dysregulation of mesenchymal cells.[23] Characteristic cutaneous manifestations include pachydermia (ie, thickening of facial skin resulting in leonine faces) over the scalp, cutis verticis gyrata, and bilateral ptosis over the eyes resulting in blepharoptosis.[24] These changes yield a characteristic “bull-dog” appearance.[25]
Other influences are acne, eczema, seborrhea, and palmoplantar hyperhidrosis. The skin of the hands and feet are also thickened, but usually not folded.
NextThe etiology of hypertrophic osteoarthropathy is unknown. Several mechanisms have been proposed as contributing to the pathophysiology of hypertrophic osteoarthropathy. Paraneoplastic growth factors[26] like prostaglandin E, other cytokines, neurologic, hormonal,[27] and immune mechanisms[20] and vascular thrombi caused by platelets and antiphospholipid antibodies[28] have all been proposed as possible etiologies.[13] All or at least many probably contribute to its development in the different clinical situations. A popular current theory involves the interaction between activated platelets and the endothelium.[26, 28, 29]
The most important of these mechanisms results from the fact that many circulating signaling molecules and growth factors are normally cleared from the blood by the pulmonary endothelium.[19] Normally, platelets are fragmented in the pulmonary microvasculature before they reach the general circulation. In 1987, Dickinson and Martin suggested that hypertrophic osteoarthropathy is related to the presence of megakaryocytes and many circulating factors normally inactivated by the lungs that have bypassed the lung circulatory network and lodged in the fingertip circulation.[30, 4] This has been been proven in patients with cyanotic heart diseases that have been found large circulating platelets with abnormal and, at times, bizarre morphology. Those macrothrombocytes are responsible for the aberrant platelet volume distribution curves.[31, 28]
To date, several physiologic and anatomic processes have been defined in which these large particles reach the fingertips and impact release of growth factors, including bypassing of megakaryocytes or megakaryocyte fragments through the lung capillary network (eg, right-to-left intracardiac shunts, carcinoma of the bronchus, anatomic malformation of the vasculature, patent ductus arteriosus complicated by pulmonary hypertension and a right-to-left shunt), formation of large platelet clumps on the left side of the heart or in large arteries (eg, subacute bacterial endocarditis, subclavian aneurysm), or chronic platelet excess (eg, chronic inflammatory bowel disease).[32]
For the reasons above, cyanotic heart diseases is an excellent model for studying hypertrophic osteoarthropathy pathogenesis because more than one third of patients with lifelong clubbing secondary to cyanotic heart disease eventually display the full hypertrophic osteoarthropathy syndrome.[33] Hypertrophic osteoarthropathy caused by intrapulmonary shunting of blood become evident only in the limbs that receive unsaturated blood, for example, in patients with patent ductus arteriosus complicated by pulmonary hypertension and a right-to-left shunt.
Having escaped fragmentation in the lung microvasculature and reached the systemic circulation, megakaryocytes or megakaryocyte fragment impaction at distal sites may lead to local endothelial cell activation through the release of growth factors ie, bradykinin, slow-reacting substance of anaphylaxis, transforming growth factor-β1 (TGF-β1), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) stored in the platelet alpha-granules. These are all angiogenic, with trophic effects on capillary beds.
In addition, they all enhance the activity of osteoblasts and fibroblasts. This initiates finger clubbing by inducing connective-tissue matrix synthesis periostosis.[28, 29] Increased circulating growth factor levels thus would explain all of the features of hypertrophic osteoarthropathy. PDGF and VEGF are thought to contribute significantly to the development of hypertrophic osteoarthropathy. VEGF is a platelet-derived factor; its action is induced by hypoxia. It is a potent angiogenic and permeability-enhancing factor, as well as a bone-forming agent. VEGF receptors are expressed in subperiosteal bone-forming cells. Both the PDGF and VEGF induce vascular hyperplasia, new bone formation and edema.[34]
In keeping with this hypothesis, Matucci-Cerinic et al have shown elevated von Willebrand factor antigen (vWF:Ag) levels in persons with primary hypertrophic osteoarthropathy and in persons with hypertrophic osteoarthropathy secondary to cyanotic heart disease.[28] vWF:Ag is a surrogate marker of endothelial activation and damage because high plasma levels of vWF:Ag are also found in the vasculitides, myocardial infarction, diabetic microangiopathy, and scleroderma.[28] Thus, a common pathogenetic pathway for hypertrophic osteoarthropathy possibly involves localized activation of endothelial cells by an abnormal platelet population. Macrothrombocyte and endothelial cell activation can also be present in cases of hypertrophic osteoarthropathy associated with other disease entities such as liver cirrhosis, in which a prominent intrapulmonary shunting of blood occurs.[29]
Stimulation of fibroblasts by PDGF, epidermal growth factor (EGF) and TGF-β along with over expression of VEGF have also been linked to extensive myelofibrosis seen in few cases of pachydermoperiostosis.[35]
Kozak et al tested the hypothesis that digital clubbing in patients with lung cancer reflects elevated systemic levels of prostaglandin E2 and found that the median urinary level of the metabolite of prostaglandin E2 was 2.3-fold higher in patients with clubbing compared with patients without clubbing (data not shown).[36]
A second proposed mechanism for the development of HPOA is a vagally-mediated alteration in limb perfusion. Interestingly, the anatomic distribution of vagal nerve fibers correlates to the area of clubbing. Vagotomy and sympatholytic drugs have been reported to reverse or to improve hypertrophic osteoarthropathy, suggesting a role for reflex vagal stimulation.[37] Bazaar and Yun proposed that sympathetic override of the normal protective function of vagal innervation is the basis of hypertrophic osteoarthropathy.[38] Sympathetic activity has been noted to induce cytokine changes consistent with inflammation.
Among these, epinephrine has been shown to induce production of interleukin (IL)-11 in human osteoblasts. Recombinant IL-11 has been shown to cause reversible symmetric periostitis in the extremities. In diseased states, autonomic stimulation may occur as a result of chemoreceptor activation in response to acidosis, hypoxia, or hypercapnia. Examples include sleep apnea, congestive heart failure, renal failure, and tumor-induced hypoxia. Removal of the associated lung neoplasm or correction of a cyanotic heart malformation has similar effects, suggesting that alteration of lung function plays an important role.[26]
A third mechanism is the possibility of ectopic production of hormonelike substances (like VEGF) by tumor or inflammatory tissue, resulting in excessive circulating levels of angiogenic substances that would cause capillary bed hypertrophy and periosteal reaction, as noted earlier.Two case reports have independently noted elevated circulating concentrations of VEGF and evidence of tumor production of VEGF associated with lung cancer. Following tumor resection, the concentrations of VEGF markedly decline, which also correlates with clinical improvement.
Diverse types of cancer growths produce VEGF as a mechanism of tumor dissemination. Abnormal expression of VEGF is known to occur in diseases associated with hypertrophic osteoarthropathy, such as mesothelioma, Graves disease and inflammatory bowel disease. These diseases are characterized by prominent endothelial cell involvement, leading to overproduction of VEGF and thus acropachy. Increase level of VEGF and IL-6 caused by the genetic mutation of K-ras might play a role in the pathogenesis of hypertrophic osteoarthropathy with lung cancer.[39]
Alonso-Bartolome et al suggested involvement of the humoral pathway giving rise to graft infection associated hypertrophic osteoarthropathy syndrome by endotoxin or vasoactive compound activated or released by bacteria adherent to the graft.[4]
Chronic activation of macrophages secondary to pulmonary pathologies may lead to digital clubbing by continual production of profibrotic tissue repair factors (eg, growth factors, fibrogenic cytokines, angiogenic factors, remodelling collagenases). These factors act systemically, but their effect is greatest at those parts of the vasculature which are most sensitive to these actions, such as the nail beds. Hypoxia also triggers the activation of macrophages.[40]
Recently, the role of different cytokines and cell receptors, including IL-6 and osteoprotegerin or RANKL system have been described on the development of the disease. Higher serum levels of IL-6 and RANKL are associated with increased values in markers of bone resorption (degradation products of C-terminal telopeptides of type-I collagen and urinary hydroxyproline/creatinine ratio) and reduced serum levels of bone alkaline phosphatase, a marker of bone formation, suggesting that hypertrophic osteoarthropathy is characterized by increased bone resorption, probably mediated by IL-6 and RANKL.[41]
Pathogenesis underlying the increase involved of males in hypertrophic osteoarthropathy is been described by Bianchi et al, which proposed high levels of nuclear steroid receptors, increased cytosolic estrogen receptors, and no detectable progesterone and androgen cytosolic receptors in hypertrophic osteoarthropathy, suggesting increased tissue sensitivity to different circulating sex steroids, which could enhance tissue epidermal growth factor or transforming growth factor alpha production and use.[41]
Hypertrophic osteoarthropathy can be associated with pregnancy and aging secondary to platelet abnormalities, hormonal disturbances, and cytokine dysfunction.
Enhanced Wnt genetic signaling contributes to the development of pachydermia skin changes in primary hypertrophic osteoarthropathy by enhancing dermal fibroblast functions.[23]
Recently, a homozygous mutation in the HPGD gene, which encodes 15-hydroxyprostaglandin dehydrogenase (15-PGDH), was found to be associated with pachydermoperiostosis. However, mutations in HPGD have not been identified in Japanese pachydermoperiostosis patients.
SLCO2A1 is a novel gene responsible for pachydermoperiostosis. Although the SLCO2A1 gene is only the second gene discovered to be associated with pachydermoperiostosis, it is likely to be a major cause of pachydermoperiostosis in the Japanese population.[42] Associations of primary hypertrophic osteoarthropathy with novel mutations in the SLCO2A1 gene in Chinese patients have also been reported.[43, 44]
United States
Primary hypertrophic osteoarthropathy is a rare condition. The association of hypertrophic osteoarthropathy with chronic lung and heart diseases was established as early as 1890.[4] No systematic prevalence studies have been performed for secondary hypertrophic osteoarthropathy, but hypertrophic osteoarthropathy is associated with many illnesses.
According to Rassam et al, the occurrence of hypertrophic osteoarthropathy in lung cancer was about 3% (9 of 280) in a consecutive series seen between 1970-1975. Other literature has described a higher prevalence in primary lung cancer of about 4–32%.[4]
In congenital cardiac disease, hypertrophic osteoarthropathy has been found in 10 of 32 patients (31%). Hypertrophic osteoarthropathy associated with respiratory failure is reported to be present in 2–7% of patients.
International
Hypertrophic osteoarthropathy likely has the same incidence and prevalence around the world.
The mortality and morbidity of hypertrophic osteoarthropathy vary with the associated illness.
PHO has a self-limiting course, and progression stops at the end of adolescence. There is no curative treatment for the skeletal abnormalities.[9]
Hypertrophic osteoarthropathy affects persons of all races.
Hypertrophic osteoarthropathy has a marked predominance in males, with a male-to-female ratio of 9:1.[45] It has an autosomal dominant pattern of inheritance, with mainly variable expression and incomplete penetrance and familial aggregation in 25-38% of cases.[7, 25] Recessive autosomal inheritance and X-linked mutations may also be present, but they may differ in severity and prevalence of clinical features.[46] Secondary osteoarthropathy has the same sex ratio as the associated illnesses.
Primary hypertrophic osteoarthropathy has a bimodal peak of onset that occurs in patients younger than 1 year and in patients who are around puberty, ie, approximately age 15 years.[45] Secondary hypertrophic osteoarthropathy is rarely encountered in children and adolescents. It most commonly affects individuals aged 55-75 years.[4]
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved