Gout and pseudogout are the 2 most common crystal-induced arthropathies. Gout (see the image below) is caused by monosodium urate monohydrate crystals; pseudogout is caused by calcium pyrophosphate crystals and is more accurately termed calcium pyrophosphate disease.
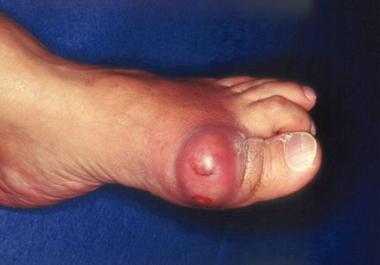 Gout. Acute podagra due to gout in elderly man.
Gout. Acute podagra due to gout in elderly man.
Symptoms of gout or pseudogout include the following:
Physical findings may include the following:
Complications of gout include the following:
See Presentation for more detail.
Studies that may be helpful include the following:
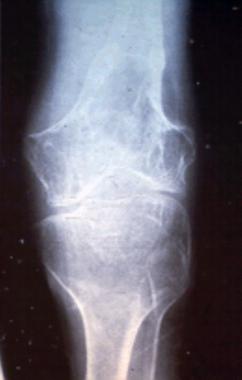
Plain radiographs may show findings consistent with gout. Erosions with overhanging edges are generally considered pathognomonic for gout (though also found in other diseases). Characteristics of erosions typical of gout include the following:
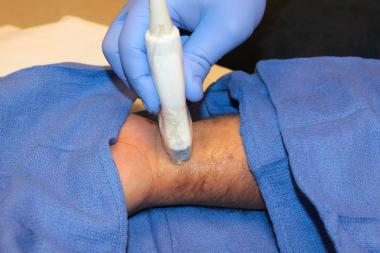
Ultrasonographic findings in established gout include the following:
Other imaging modalities that may be considered include the following:
See Workup for more detail.
Gout is managed in the following 3 stages:
Acute treatment of proven crystal-induced arthritis is directed at relief of the pain and inflammation. Agents used in this setting include the following:
Therapy to control the underlying hyperuricemia generally is contraindicated until the acute attack is controlled (unless kidneys are at risk because of an unusually heavy uric acid load).
Long-term management of gout is focused on lowering uric acid levels. Agents used include the following:
Because these agents change serum and tissue uric acid levels, they may precipitate acute attacks of gout. This undesired effect may be reduced by prophylaxis with the following:
Other therapeutic agents that may be considered include the following:
Nonpharmacologic measures that may be warranted are as follows:
See Treatment and Medication for more detail.
NextGout and pseudogout are the two most common crystal-induced arthropathies. Gout is caused by monosodium urate monohydrate crystals; pseudogout is caused by calcium pyrophosphate (CPP) crystals and is more accurately termed calcium pyrophosphate disease (CPPD). (See Pathophysiology and Etiology.) Gout is one of the oldest diseases in the medical literature,[1, 2] known since the time of the ancient Greeks. Pseudogout, which may be clinically indistinguishable from gout, was recognized as a distinct disease entity in 1962.
Crystal deposition can be asymptomatic, but gout and CPPD can develop into debilitating illnesses marked by recurrent episodes of pain and joint inflammation that result from the formation of crystals within the joint space and deposition of crystals in soft tissue.[3, 4, 5] If untreated, these disorders can lead to joint destruction and, in the case of uric acid crystals, renal damage.
Elevated serum uric acid levels are the principal risk factor for developing gout. lIn study that compared 993 patients with asymptomatic hyperuricemia and 4,241 normouricemic patients, the odds ratio (OR) for developing gout was 32 times higher in the hyperuricemic group than in the normouricemic group. The risk was most striking in men with severe hyperuricemia, in whom the OR for developing gout was 624.8.[6]
Although gout is associated with hyperuricemia, gout attacks are triggered not by a particular level of uric acid but typically by acute changes in the level of uric acid. All individuals with gout have hyperuricemia; however, hyperuricemia is also found in patients taking diuretics and even in those taking niacin or low doses of aspirin.
Gout may be either primary or secondary (see Etiology). Primary gout is related to underexcretion or overproduction of uric acid, often associated with a mix of dietary excesses or alcohol overuse and metabolic syndrome. Secondary gout is related to medications or conditions that cause hyperuricemia, such as the following[7] :
Gout is definitively diagnosed on the basis of demonstration of urate crystals in aspirated synovial fluid, in the absence of another etiology for arthritis. Classic radiographic findings are highly suggestive (see Workup).
Advances in early diagnosis and the availability of definitive treatment have significantly improved the prognosis of gout, as evidenced by the declining incidence of disabling chronic tophaceous gout. However, tophaceous gout may still develop because of misdiagnosis, poor management, medication intolerances, or poor patient adherence.
Gout is managed in the following 3 stages:
Treatment of gout is important to relieve pain; to prevent disease progression; and to prevent deposition of urate crystals in the renal medulla or uric acid crystals in the renal collecting system, which may produce kidney stones or urate nephropathy.[8] (See Treatment.)
Management of pseudogout also involves treatment of the acute attack and prophylaxis. Treatment of the acute phase of pseudogout follows the same approaches as are used in gout, and colchicine is effective for prophylaxis. In contrast with gout, however, no specific therapeutic regimen exists to treat the underlying cause of CPP crystal deposition in pseudogout, except in cases associated with disorders such as hemochromatosis or hyperparathyroidism. (See Treatment.)
Gout can be considered a disorder of metabolism that allows uric acid or urate to accumulate in blood and tissues. When tissues become supersaturated, the urate salts precipitate, forming crystals. In addition, the crystals also are less soluble under acid conditions and at low temperatures, such as occur in cool, peripheral joints (eg, the metatarsophalangeal joint of the big toe).
Urate initially precipitates in the form of needlelike crystals. The light-retarding (phase-shifting) characteristics of urate crystals allow them to be recognized by polarizing microscopy (see the image below).
 Gout. Needles of urate crystals seen on polarizing microscopy.
Gout. Needles of urate crystals seen on polarizing microscopy.
Many conditions and drugs have been associated with an increase in plasma (and subsequent synovial) urate levels, particularly metabolic syndrome. A genetic predisposition for hyperuricemia exists; except in rare genetic disorders, however, the development of gout in hyperuricemic individuals appears to be mediated by environmental factors.[9, 10, 11]
The CPP crystals that produce pseudogout comprise a combination of inorganic pyrophosphate and calcium. The inorganic pyrophosphate is produced in large part by ectonucleotide phosphodiesterase pyrophosphatase (ENPP1), a catalytic enzyme found in chondrocytes of cartilage, and the pyrophosphate is exported potently by the membrane transporter ANKH.
A genetic predisposition exists for pseudogout. However, aging, some metabolic diseases (eg, hyperparathyroidism, hemochromatosis, and hypomagnesemia), and any process that leads to osteoarthritis also can be associated with subsequent CPP crystal deposition and pseudogout.
The presence of urate crystals in the soft tissues and synovial tissues is a prerequisite for a gouty attack. However, these crystals can also be found in synovial fluid or on the cartilage surface in the absence of joint inflammation.
A gout attack may be triggered either by release of crystals (eg, from partial dissolution of a microtophus caused by changing serum urate levels) or by precipitation of crystals in a supersaturated microenvironment (eg, release of urate as a consequence of cellular damage). In either situation, it is believed, naked urate crystals then interact with intracellular and surface receptors of local dendritic cells and macrophages, triggering a danger signal to activate the innate immune system.[12]
This interaction may be enhanced by immunoglobulin G (IgG) binding.[13, 14] Triggering of these receptors, including Toll-like receptors, followed by intracellular signaling by the NLRP3 inflammasome, results in the release of interleukin (IL)-1β, which in turn initiates a cascade of proinflammatory cytokines, including IL-6, IL-8, neutrophil chemotactic factors, and tumor necrosis factor (TNF)-α.[15, 16] Neutrophil phagocytosis leads to another burst of inflammatory mediator production.
Subsidence of an acute gout attack results from multiple mechanisms, including the clearance of damaged neutrophils, change in the properties of urate crystals, and the production of anti-inflammatory cytokines such as IL-1 receptor antagonist (IL-1RA), IL-10, and transforming growth factor (TGF)-β.[14, 17, 18, 19]
Gout develops in the setting of excessive stores of uric acid in the form of monosodium urate. Uric acid is an end-stage by-product of purine metabolism. Humans remove uric acid primarily by renal excretion. When excretion is insufficient to maintain serum urate levels below the saturation level of 6.8 mg/dL, hyperuricemia may develop, and urate can crystallize and deposit in soft tissues.
About 90% of patients with gout develop excess urate stores because of an inability to excrete sufficient amounts of uric acid in the urine (underexcretion). Most of the remaining patients either overconsume purines or produce excessive amounts of uric acid endogenously (overproduction). A few have impaired intestinal elimination of uric acid.
In rare cases, overproduction of uric acid is the result of a genetic disorder, such as the following[20] :
Overproduction of uric acid may also occur in disorders that cause high cell turnover with release of purines that are present in high concentration in cell nuclei. These disorders include myeloproliferative and lymphoproliferative disorders, psoriasis, and hemolytic anemias. Cell lysis from chemotherapy for malignancies, especially those of the hematopoietic or lymphatic systems, can raise uric acid levels, as can excessive exercise and obesity.
Causes of secondary gout due to underexcretion of uric acid include renal insufficiency, lead nephropathy (saturnine gout), starvation or dehydration, certain drugs, and chronic abuse of ethanol (especially beer and hard liquor). These disorders should be identified and corrected, if possible.
Certain comorbid conditions are associated with a higher incidence of gout, including the following[21, 22] :
Foods that are rich in purines include anchovies, sardines, sweetbreads, kidney, liver, and meat extracts. Consumption of fructose-rich foods and beverages (eg, those sweetened with high-fructose corn syrup) is associated with an increased risk of gout in both men and women.[23, 24]
The heritability of serum urate levels is estimated at 63%.[25] Genome-wide association studies (GWAS) have identified several candidate loci associated with chronically elevated serum urate concentrations and gout.[26, 27, 28, 29]
In particular, 3 genes are noted to have a strong association with hyperuricemia. The locus with the strongest evidence of association is the glucose transporter 9 (GLUT9) gene, commonly referred to as the solute carrier 2A9 (SLC2A9), the product of which alters the renal excretion of uric acid. Some of the variants are associated with a protective effect, whereas others convey a higher risk of gout.[30]
The urate transporter 1 (URAT1) gene is involved with the urate-organic anion exchanger. Several mutations in this gene have been associated with gout.
Polymorphisms in the ABCG2 gene, which is located on chromosome 4 and codes for an intestinal urate transporter, are strongly associated with high serum uric acid concentrations and gout. Elevation of uric acid levels is greater in men than in women with the minor allele of rs2231142 in ABCG2.[26, 28]
Although genetic factors have been strongly associated with hyperuricemia, environmental and other state-of-health factors are responsible for the majority of the gout burden in developed countries.[30, 31] A study of 514 male twin pairs did show a strong concordance in hyperuricemia among monozygotic (MZ) twins (53%) as compared with dizygotic (DZ) twins (24%), but it did not show a significant difference between MZ and DZ twins with regard to the lifetime prevalence of gout.[11]
Individual gout flares are often triggered by acute increases or decreases in urate levels that may lead to the production, exposure, or shedding of crystals. Changes in urate levels can result from acute alcohol ingestion, acute overindulgence in foods high in purines, rapid weight loss, dehydration, or trauma.
Similarly, flares can be precipitated by additions of or changes in dosage of medications that raise or lower uric acid levels. Medications that increase uric acid levels via effects on renal tubular transport include loop and thiazide diuretics, niacin, low-dose aspirin, and cyclosporine A.[32, 33, 34] Agents that lower levels of uric acid include radiocontrast dyes, xanthine oxidase inhibitors (eg, allopurinol and febuxostat), and uricosurics (eg, probenecid).
Although the pathophysiology, clinical presentation, and acute-phase treatment of gout and pseudogout are very similar, the underlying causes of the 2 diseases are very different. Many cases of pseudogout in elderly people are idiopathic, but pseudogout has also been associated with trauma and with many different metabolic abnormalities, the most common of which are hyperparathyroidism and hemochromatosis.
Risk factors for pseudogout include use of loop diuretics (but not thiazide diuretics) and proton pump inhibitors, which cause hypomagnesemia.[34] Pseudogout attacks have been reportedly induced by etidronate disodium therapy and angiography.[35, 36]
Pseudogout has been recognized as having an underlying genetic component; however, comorbid conditions (such as osteoarthritis) and environmental factors are thought to play a much stronger role.[37] Some disorders that can lead to secondary pseudogout, such as hemochromatosis, do have a clear genetic cause. These patients should be properly evaluated and counseled.
Gout affects 8.3 million people in the United States; prevalence among adults is estimated to be 3.9%, on the basis of data from the 2007-2008 National Health and Nutrition Examination Survey (NHANES).[38] Prevalence is approximately 20% in patients with a family history of gout. It is estimated that more than 2 million people in the United States take medication to decrease serum uric acid levels.
Gout has become increasingly common in the United States as the population has grown older and heavier.[39] From 1990 to 1999, the incidence rose 40%.[40] Estimates for the number of US adults with self-reported gout in the previous year rose from 2.1 million in 1995 to 3 million in 2008.[10] In 2008, gout accounted for 174,823 emergency department (ED) visits in the US, or approximately 0.2% of all ED visits.[41]
The frequency of pseudogout varies with age. The annual incidence of acute attacks of arthritic pain and swelling is about 1.3 per 1000 adults, but nearly 50% of adults develop radiographic changes typical of CPPD by age 80 years.
Attacks of gout have been noted to occur more frequently in the spring and less frequently in the winter. The reason for this is unknown.
Gout has a worldwide distribution. The prevalence varies widely from country to country. Regional differences may reflect environmental, dietary, and genetic influences.[42]
In the United Kingdom from 2000 to 2007, the incidence of gout was 2.68 per 1000 person-years—4.42 in men and 1.32 in women, and increasing with advancing age.[43] In Italy, the prevalence of gout rose from 6.7 per 1000 population in 2005 to 9.1 per 1000 population in 2009, increasing with age and 4 times higher in men.[44] In the Maori people of New Zealand, studies from the 1970s found that 0.3% of men and 4.3% of women were affected.[45, 46]
Gout has a male predominance.[24, 47] The estimated prevalence of gout is 5.9% in men and 2% in women.[38] This difference is largely a consequence of age at onset; estrogenic hormones have a mild uricosuric effect, and gout is therefore unusual in premenopausal women. For pseudogout, the male-to-female ratio is approximately 50:50.
The predominant age range of gout is 30-60 years. Usually, uric acid levels are elevated for 10-20 years before the onset of gout. In men, uric acid levels rise at puberty, and the peak age of onset of gout in men is in the fourth to sixth decade of life. However, onset may occur in men in their early 20s who have a genetic predisposition and lifestyle risk factors.[48] In women, uric acid levels rise at menopause, and peak age of onset is in the sixth to eighth decade of life.
The rate of gout is almost 5 times higher in persons aged 70-79 years than in those younger than 50 years.[49] The higher prevalence of gout in elderly persons may also reflect an increased prevalence of metabolic syndrome, high rates of diuretic treatment for hypertension and chronic heart failure, and the use of low-dose aspirin.[50]
Earlier onset of gout occurs in patients with renal insufficiency or a genetic abnormality of purine metabolism (eg, hypoxanthine-guanine phosphoribosyltransferase deficiency or phosphoribosylpyrophosphate synthetase superactivity). Cyclosporine A can cause an accelerated form of gout, even in premenopausal women, that can present after only a few years of hyperuricemia, particularly if the patient is also receiving diuretics.
Gout has an increased prevalence in some populations but is rare in others. For example, the frequency of gout is higher in populations such as the Chamorros and Maori and in the Blackfoot and Pima tribes. Many Maori and other Polynesian women have a genetic defect in renal urate handling that places them at risk for hyperuricemia and gout.[51] However, racial differences may at least in part reflect differences in diet, which has a large influence on the clinical expression of gout.
In the United States, the incidence of gout is 3.11 per 1000 person-years in African Americans and 1.82 per 1000 person-years in whites; the excess risk can be partly explained by a higher frequency of incident hypertension.[52] In contrast, clinically recognized gout is extremely rare among blacks living in Africa.[53]
Gout is associated with considerable morbidity, with acute episodes often causing incapacitation. However, gout that is treated early and properly carries an excellent prognosis if patient adherence to treatment is good.
With early treatment, gout should be totally controlled. If attacks recur, successful uric acid adjustment (requiring lifelong use of urate-lowering medication) usually suppresses further activity. During the first 6-24 months of urate-lowering therapy, acute attacks of gout often occur more frequently.[54, 55]
Chronic injury to intra-articular cartilage leaves the joints more susceptible to subsequent joint infections. Draining tophi can become secondarily infected. Untreated chronic tophaceous gout can lead to severe joint destruction and, rarely, renal impairment. Deposition of monosodium urate crystals in the kidney can result in inflammation and fibrosis, leading to reduced renal function or chronic nephropathy.[56] Rarely, gout can produce spinal cord impingement when deposition in tissues produces a local mass.
Acute attacks of pseudogout usually resolve within 10 days. Prognosis for resolutions of acute attacks is excellent. Some patients experience progressive joint damage with functional limitation. CPPD also can cause chronic arthritis that can resemble osteoarthritis or rheumatoid arthritis.
Hyperuricemia and gout are associated with an increased overall likelihood of mortality. Whether this is directly attributable to hyperuricemia or gout or to gout-associated diseases (eg, insulin resistance, type 2 diabetes mellitus, abdominal obesity, hypercholesterolemia, or hypertension) has been much debated.[57, 58, 59]
Although no evidence has shown that gout or hyperuricemia causes any of these disorders, elevated urate levels have been shown to correlate with elevated blood pressure in adolescents.[60] Among middle-aged men, hyperuricemia is a significant independent risk factor for death from cardiovascular disease.[61] A meta-analysis found an independent association between gout and cardiovascular mortality as well as all-cause mortality.[59]
In a 2010 study, Kuo et al demonstrated that gout, but not hyperuricemia, is associated with higher risk of death from all causes and cardiovascular diseases. Analysis of 1383 deaths among 61,527 Taiwanese subjects showed in individuals with gout compared with those who had normal uric acid levels, the hazard ratio (HR) of all-cause mortality was 1.46 and the adjusted HR of cardiovascular mortality was 1.97. Among individuals with hyperuricemia, the HR of all-cause mortality was 1.07 and the adjusted HR of cardiovascular mortality was 1.08.[62]
An analysis of nationwide data on more than 200,000 English patients indicates that individuals with gout are at increased risk for both heart attack and stroke. The rate ratio for myocardial infarction in patients with gout was 1.82. Rate ratios for stroke were 1.71 for all stroke, 1.68 for ischemic stroke, 1.69 for hemorrhagic stroke, and 2.00 for stroke of unspecified type. Risks were elevated in both men and women and were higher in the younger age groups.[63, 64]
Risk for vascular disease is increased in patients with gout, particularly women, according to a retrospective cohort study from the United Kingdom that included 8386 patients with gout and 39,766 matched controls. Multivariate analysis showed that women with gout had a 25% increased risk for any vascular event compared with women without gout (hazard ratio [HR], 1.25) and increased risks for any coronary heart disease (HR, 1.25) and peripheral vascular disease (HR, 1.89).[65, 66]
Men with gout, compared with those without gout, had a small but significantly increased risk for any vascular event (hazard ratio [HR], 1.06) and an increased risk for any coronary heart disease (HR, 1.08) and peripheral vascular disease (HR, 1.18). Unlike women, men with gout were not at greater risk for angina, transient ischemic attack, or stroke.[65, 66]
Patients with severe hyperuricemia should avoid foods with high purine content. Moderation in food and alcohol consumption is advised. Early recognition of acute gout attacks is critical, in that intervention with medication is much more effective earlier in the attack.
For patient education information, see the Arthritis Center, as well as Gout. Online information and pamphlets on gout are also available from the Arthritis Foundation.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved