Antiphospholipid syndrome (APS) is an acquired autoimmune disorder that manifests clinically as recurrent venous or arterial thrombosis and/or fetal loss.[1] Characteristic laboratory abnormalities in APS include persistently elevated levels of antibodies directed against membrane anionic phospholipids (ie, anticardiolipin [aCL] antibody, antiphosphatidylserine) or their associated plasma proteins, predominantly beta-2 glycoprotein I (apolipoprotein H); or evidence of a circulating anticoagulant.
Multiple terms for APS exist. Unfortunately, some synonyms can be confusing. Lupus anticoagulant (LA) syndrome, for example, is misleading because patients with APS may not necessarily have systemic lupus erythematosus (SLE) and LA is associated with thrombotic rather than hemorrhagic complications. In an attempt to avoid further confusion, APS is currently the preferred term for the clinical syndrome (as described below).
Some patients with APS have no evidence of any definable associated disease, while, in other patients, APS occurs in association with SLE or another rheumatic or autoimmune disorder. Traditionally, these have been referred to as primary or secondary APS, respectively. Currently, however, the preferred terminology is APS with or without associated rheumatic disease. Although antiphospholipid (aPL) antibodies are clinically linked to APS, whether they are involved in the pathogenesis or are an epiphenomenon is unclear. (Up to 5% of healthy individuals are known to have aPL antibodies.)
NextIn APS, the homeostatic regulation of blood coagulation is altered; however, the mechanisms of thrombosis are not yet defined. One hypothesis postulates a defect in cellular apoptosis, which exposes membrane phospholipids to the binding of various plasma proteins, such as beta-2 glycoprotein I. Once bound, a phospholipid-protein complex is formed and a neoepitope is uncovered, which subsequently becomes the target of autoantibodies. Recent evidence suggests that oxidized beta-2 glycoprotein I is able to bind to and activate dendritic cells in a manner similar to activation triggered by Toll-like receptor 4 (TLR-4), which could amplify the production of autoantibodies.{ref1-INVALID REFERENCE}{ref2-INVALID REFERENCE}[2]
Other proposed mechanisms for the hypercoagulable effect of aPL antibodies, which may or may not depend on beta-2 glycoprotein I, include the following:
Complement activation has been increasingly recognized as a possible significant role in the pathogenesis of APS. Emerging evidence from murine models suggests that APL-mediated complement activation may be a primary event in pregnancy loss.[2, 3]
Clinically, the series of events that leads to hypercoagulability and recurrent thrombosis can affect virtually any organ system, including the following:
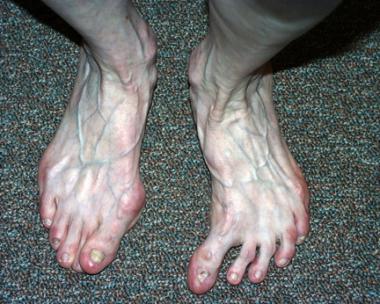
The kidney is a major target organ in APS. Nephropathy in APS is characterized by small-vessel vaso-occlusive lesions associated with fibrous intimal hyperplasia of interlobular arteries, recanalizing thrombi in arteries and arterioles, and focal atrophy.[4]
A “two-hit” theory has been proposed in which a second risk factor (age, hypertension, diabetes, obesity, smoking, pregnancy, surgery, other genetic hypercoagulable state) incites the thrombotic effects of aPL.[5]
United States
The actual frequency of APS in the general population is unknown. One to 5% of healthy individuals have aPL antibodies. It is estimated that the incidence of APS is approximately 5 cases per 100,000 persons per year, and the prevalence is approximately 40-50 cases per 100, 000 persons.[6] aCL antibodies tend to be found more frequently in elderly persons; thus, positive titer results should be interpreted with caution in this population. aPL antibodies are found in approximately 30-40% of patients with SLE, but only about 10% have APS.[7] Approximately half of APS cases are not associated with another rheumatic disease. In a study of 100 patients with confirmed venous thrombosis and no history of SLE, aCL antibodies were found in 24% and LA in 4%.
aPL syndrome is the cause of 14% of all strokes, 11% of myocardial infarctions, 10% of deep vein thromboses, 6% of pregnancy morbidity, and 9% of pregnancy losses.[8]
International
International frequency is probably similar to US frequency.
APS may contribute to an increased frequency of stroke or MI, especially in younger individuals. Strokes may develop secondary to in situ thrombosis or embolization that originates from the valvular lesions of Libman-Sacks (sterile) endocarditis, which may be seen in patients with APS. Cardiac valvular disease may be severe enough to require valve replacement. Recurrent pulmonary emboli or thrombosis can lead to life-threatening pulmonary hypertension.
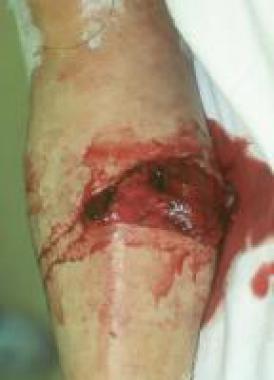
Catastrophic APS (CAPS) is a rare, serious, and often fatal manifestation (mortality rate of approximately 50%) characterized by multiorgan infarctions over a period of days to weeks.
Late spontaneous fetal loss (second or third trimester) is common; however, it can occur at any time during pregnancy. Recurrent early fetal loss (< 10 weeks’ gestation) is also possible.
No defined racial predominance for primary APS has been documented, although SLE is more common in African American and Hispanic populations.
A female predominance has been documented, particularly for secondary APS. This parallels the association of APS with SLE and other connective-tissue diseases, which also have a female predominance.
APS is more common in young to middle-aged adults; however, it also manifests in children and elderly people. Disease onset has been reported in children as young as 8 months. In an international registry of pediatric APS cases, patients without associated rheumatic disease were younger and had a higher frequency of arterial thrombotic events, whereas patients with associated rheumatic disease were older and had a higher frequency of venous thrombotic events associated with hematologic and skin manifestations.[9]
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved