In 1912, Maurice Klippel and Andre Feil independently provided the first descriptions of Klippel-Feil syndrome. They described patients who had a short, webbed neck; decreased range of motion (ROM) in the cervical spine; and a low hairline. Feil subsequently classified the syndrome into the following three categories:
Since their original description, other classification systems have been advocated to describe the anomalies, predict the potential problems, and guide treatment decisions.
In a series of articles, Samartzis et al suggested their own classification system,[1, 2] which stratified patients as follows:
Using their system, the investigators reviewed a series of patients to clarify prognosis.[1, 2]
Gray et al[3] described 462 patients with Klippel-Feil syndrome who had low hairline, short neck, and decreased range of motion (ROM). They found that the level of fusion did not greatly affect the incidence of neurologic symptoms. The most frequent level they identified was a defect of the occiput to C1, C2, and C3. These produced the most symptoms; lesions below C3 and 4 were slightly less likely to cause symptoms. Twenty-seven percent of symptoms occurred in the first decade.
Nagib et al[4] described three types and related the incidence of neurologic symptoms to each type as follows:
Patients with Klippel-Feil syndrome usually present with the disease during childhood, but they sometimes present later in life. The challenge to the clinician is to recognize the associated anomalies that can occur with Klippel-Feil syndrome and to perform the appropriate workup for diagnosis.
NextThe true incidence of Klippel-Feil syndrome is unknown: No one has ever studied a cross-section of healthy people to determine the true incidence.
Two studies investigated the incidence of Klippel-Feil syndrome, using two different means. Gjorup and Gjorup reviewed all of the radiographic cervical spine films from a single hospital in Copenhagen.[6] From these films, they determined an incidence of 0.2 cases per 1000 people. Brown et al reviewed 1400 skeletons from the Terry collection, which at that time was located at the Washington University School of Medicine.[7] They found an incidence of 0.71%.
The etiology of Klippel-Feil syndrome and its associated conditions is unknown. The syndrome can present with a variety of other clinical syndromes, including fetal alcohol syndrome, Goldenhar syndrome, and anomalies of the extremities.[8, 9, 10]
Gunderson suggested that Klippel-Feil syndrome is a genetic condition, whereas Gray found a low incidence of inheritance.[11, 12] Two small studies suggested that mutations of the MEOX1 gene, which codes for mesenchyme homeobox 1, may cause a recessive subtype of the syndrome.[13, 14]
Other investigators have considered Klippel-Feil syndrome to be some type of global fetal insult, which could explain the other associated conditions. Some have considered it to be a consequence of vascular disruption.[15, 16]
The clinical presentation of Klippel-Feil syndrome is varied because of the different associated syndromes and anomalies that can occur in patients with this syndrome. A complete history and careful physical examination may reveal some associated anomalies. From an orthopedic standpoint, most of the workup involves imaging (see Workup, Imaging Studies).
Klippel-Feil syndrome is detected throughout life, often as an incidental finding. Patients with upper cervical spine involvement tend to present at an earlier age than those whose involvement is lower in the cervical spine. Most patients present with a short neck and a decreased cervical ROM, with a low hairline occurring in 40-50% of patients. Decreased ROM is the most frequent clinical finding. Rotational loss usually is more pronounced than is the loss of flexion and extension.
Other patients present with torticollis or facial asymmetry. In very young children, it is important to differentiate congenital muscular torticollis from Klippel-Feil syndrome. It is often difficult to obtain good plain radiographs of young children with torticollis, especially of the craniocervical junction. Neurologic problems may develop in 20% of patients. Gray found that 27% developed symptoms in the first decade.[3]
Rouvreau found that five of 19 patients with Klippel-Feil syndrome had neurologic involvement; of these five, two had neurologic problems resulting from hypermobility at one level.[17] Occipitocervical abnormalities were the most common cause of neurologic problems (see the images below). Some patients present with pain.[18]
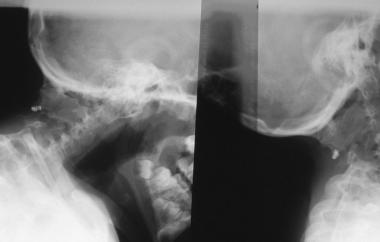 An anomaly of the occipitocervical junction in a patient with Klippel-Feil syndrome. The anomaly was unstable and was fused.
An anomaly of the occipitocervical junction in a patient with Klippel-Feil syndrome. The anomaly was unstable and was fused.
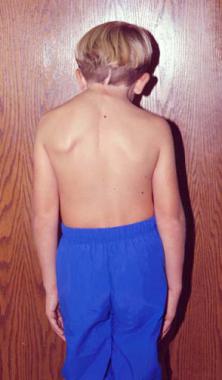 Posterior photo of a patient with Klippel-Feil syndrome and an anomaly of the occipitocervical junction. The image shows an elevated left shoulder due to a Sprengel anomaly; a short, webbed neck; and a low hairline.
Posterior photo of a patient with Klippel-Feil syndrome and an anomaly of the occipitocervical junction. The image shows an elevated left shoulder due to a Sprengel anomaly; a short, webbed neck; and a low hairline.
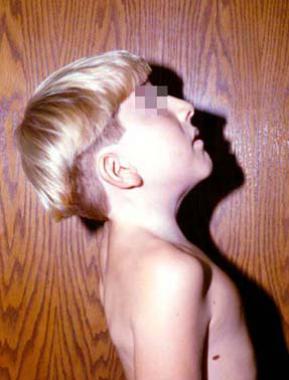 This patient has Klippel-Feil syndrome and an anomaly of the occipitocervical junction. The patient's flexion and extension after the occipitocervical fusion is demonstrated. His rotation was very limited.
This patient has Klippel-Feil syndrome and an anomaly of the occipitocervical junction. The patient's flexion and extension after the occipitocervical fusion is demonstrated. His rotation was very limited.
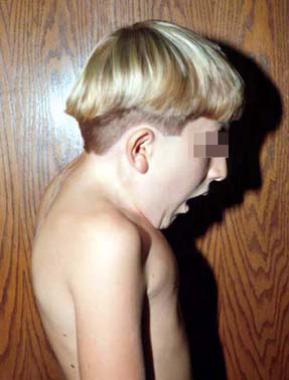 Flexion of the cervical spine in a patient who had an occipitocervical fusion.
Flexion of the cervical spine in a patient who had an occipitocervical fusion.
Nagib et al[4] reviewed 21 cases of Klippel-Feil syndrome (10 male, 11 female) over a 25-year period to identify high-risk patients and describe treatments required. Eight patients had been admitted for genitourinary complications or cardiovascular or otologic anomalies. Of the 21, 12 had no neurologic deficit, and 11 of these 12 had a single block vertebra with no other cervical or craniocervical anomalies; nine had neurologic deficits occurring spontaneously or after minor trauma (these had the most varied and complicated radiographic findings).
According to the classification system described above, there were four type I patients with an unstable fusion pattern, three type II patients with craniocervical abnormalities, and two type III patients with fusion anomalies and spinal stenosis.[4] Nine patients (43%) required decompression and stabilization in the second or third decade of life.
The investigators concluded that types I and II are commonly combined with hypermobility of the craniocervical junction.[4] Foramen magnum encroachment may be associated with tight dural bands or upward migration of the odontoid. In type III patients, they found that the level of stenosis could be above or below the abnormal area.
Hensinger et al, in a review of 50 patients with Klippel-Feil syndrome, found that 30 (60%) had associated scoliosis.[19, 20] In some patients with Klippel-Feil syndrome, the scoliosis is congenital (see the image below), owing to the involvement of other parts of the thoracic or lumbar spine. Other patients develop scoliosis in the thoracic spine, to compensate for cervical or cervicothoracic scoliosis. In addition to fusion anomalies in the cervical spine, cervical spinal stenosis can occur; though uncommon, it can increase the risk of neurologic involvement.
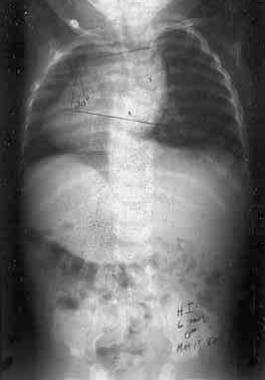 This anteroposterior radiograph of the spine in a patient with Klippel-Feil syndrome demonstrates congenital scoliosis and a Sprengel deformity.
This anteroposterior radiograph of the spine in a patient with Klippel-Feil syndrome demonstrates congenital scoliosis and a Sprengel deformity.
Anomalies of the craniocervical junction can cause instability at lower segments. Traumatic tetraplegia has been reported after minor trauma.[21] A Sprengel anomaly occurs in 20-30% of patients.[22] The ROM of the shoulders must be checked, and the patient should be examined for an omovertebral bone, an osteocartilaginous connection that tethers the scapula to the spine (see the image below).
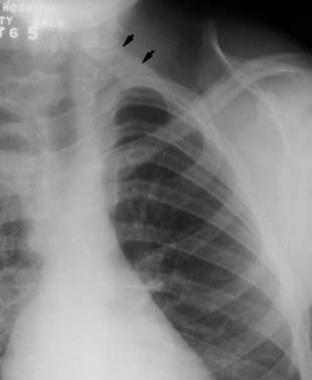 This radiograph demonstrates an omovertebral bone (marked with 2 arrows). This anomaly limits cervical spine motion.
This radiograph demonstrates an omovertebral bone (marked with 2 arrows). This anomaly limits cervical spine motion.
An omovertebral bone ossifies with age, further limiting the ROM. Computed tomography (CT) is best for demonstrating the presence of an omovertebral bone; however, this feature can also be detected through palpation or radiography. Other upper-extremity anomalies occur less frequently. A thorough examination of the ROM and function of the upper extremity must be performed.
Renal anomalies are common in individuals with Klippel-Feil syndrome, and they can be quite serious. Out of 41 patients in Hensinger's series who underwent intravenous pyelography (IVP), 16 were found to have renal anomalies. Minor renal anomalies—including a double collecting system, renal ectopia, and bilateral tubular ectasia—were detected in six of these individuals. Major renal anomalies—including hydronephrosis, absence of a kidney (see the image below), and a horseshoe kidney —were detected in 10.
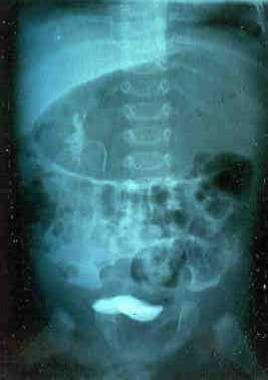 This intravenous pyelogram was performed before ultrasound was available to image the kidneys. Note unilateral absence of the left kidney.
This intravenous pyelogram was performed before ultrasound was available to image the kidneys. Note unilateral absence of the left kidney.
For patients with Klippel-Feil syndrome, ultrasonography (US) scanning now serves as the initial test to determine whether both kidneys are functioning.[23]
Cardiovascular anomalies, mainly septal defects, were found in seven patients in Hensinger's series, with four of these individuals requiring corrective surgery. Synkinesia, or mirror movement (see the image below), occurred in nine of the 50 patients. Hearing was impaired in 15 of 41 patients tested. Early audiometric and otologic evaluation are indicated in all children when the diagnosis of Klippel-Feil syndrome is established.[24]
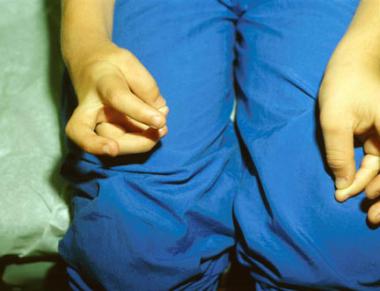 This photo demonstrates synkinesia. As the patient attempts to oppose the thumb and finger of the right hand, the same movement occurs involuntarily in the left.
This photo demonstrates synkinesia. As the patient attempts to oppose the thumb and finger of the right hand, the same movement occurs involuntarily in the left.
Torticollis and facial asymmetry occur in 21-50% of patients with Klippel-Feil syndrome. These persons may also have a muscular torticollis.[25] Craniofacial anomalies can occur as well.[26]
Less common anomalies associated with Klippel-Feil syndrome include congenital limb deficiencies, craniosynostosis, ear abnormalities, iniencephaly, and craniofacial abnormalities.[27, 28, 29, 30, 31, 32] (See the image below.)
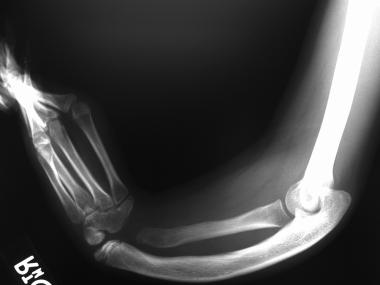 Congenital anomaly of the forearm in a patient with Klippel-Feil syndrome.
Congenital anomaly of the forearm in a patient with Klippel-Feil syndrome.
Patients with Klippel-Feil syndrome present at different ages with varying clinical manifestations. Indications for workup vary individually. For the orthopedic surgeon, the most frequent indications for surgery depend on the amount of deformity, its location, and its progression with time. Other indications include instability of the cervical spine and/or neurologic problems. These indications can occur with craniocervical junction anomalies and when two fused segments are separated by a normal segment.
Some patients present early in life with complex cervical and cervicothoracic deformity that is progressive and disfiguring. Some of these patients require cervical spine fusions to prevent progression.
Other patients may develop compensatory or associated congenital scoliosis, which also can be progressive over time and requires fusion to prevent progressive deformity. More than 50% of the patients in Hensinger's study had scoliosis.[19] Treatment of the scoliosis with bracing or surgery was required in 18 of the 50 patients.
Using their own classification system, Samartzis et al reviewed 28 patients radiographically and clinically (mean age at presentation, 7.1 years; mean age of symptom onset in symptomatic patients, 11.9 years; mean follow-up, 8.5 years).[2] Of the 28, 64% had no symptoms, two developed myelopathic symptoms (type II and type III), and two developed radiculopathic symptoms (type II and type III). Axial symptoms were more common in type I patients. The investigators recommended activity modification in high-risk patients.
The same authors reported on a patient who developed a symptomatic cervical disc herniation.[1] The patient had occipitalization of C1 and fusion of C2-3 and C4-T1. This left only C3-4 as a hypermobile segment; thus, the patient was at high risk. The patient was treated successfully with a same-day, combined anteroposterior (AP) procedure.
Theiss et al reviewed 32 patients with congenital scoliosis followed for more than 10 years.[33] Only seven (22%) developed cervical or cervical-related symptoms, and only two required surgery for their cervical-related symptoms. No fusion pattern was identified that placed the patients at greater risk for developing symptoms.
Auerbach et al studied spinal cord dimensions in children with Klippel-Feil syndrome.[34] They reviewed magnetic resonance imaging studies and clinical records of Klippel-Feil patients and age-matched controls. Torg ratios were measured, and the Torg-Pavlov ratios were found to be identical in the two groups.
The cross-sectional area of the spinal cord was smaller in Klippel-Feil syndrome patients at each level from C2-C7.[34] These differences were statistically significant, with no differences in the cerebrospinal fluid (CSF) column, suggesting that the cord size is smaller in children with Klippel-Feil syndrome than in control subjects. Four of the 12 children with Klippel-Feil syndrome presented with neurologic symptoms that improved after posterior cervical stabilization.
Samartzis et al studied the extent of fusion in the congenital K-F segment to evaluate the presence and extent of specific fusion patterns across the involved cervical segments.[35] In older patients, complete fusion was more prevalent in regard to C2-C7. In the absence of complete fusion, fusion of the posterior elements was noted more often than fusion of the anterior elements.
In another paper, Samartzis et al reviewed the role of the congenitally fused segments in 29 Klippel-Feil syndrome patients in relation to the space available to the cord (SAC) and associated cervical spine-related symptoms (CSS).[36] They suggested that an arrest of normal vertebral development may affect appositional bone development. The effect on vertebral body width may delay neurologic compromise resulting from the congenital fusion process and subsequent degenerative manifestations.
Because Klippel-Feil syndrome is associated with a constellation of possible abnormalities, no set of definite contraindications exists. If a surgeon believes that an operation is indicated, it is incumbent upon him or her to make certain that none of the conditions that could cause morbidity or mortality are present.
Cervical or occipitocervical instability could increase the risk of neurologic damage during intubation. An underlying heart defect could increase anesthetic risk. An underlying spinal stenosis or spinal cord abnormality could increase the risk of neurologic damage during spinal fusion for correction of deformity. A thorough workup of the patient is imperative prior to surgical intervention.
Workup
Copyright © www.orthopaedics.win Bone Health All Rights Reserved