Muscular dystrophy (MD) is a collective group of inherited noninflammatory but progressive muscle disorders without a central or peripheral nerve abnormality. The disease affects the muscles with definite fiber degeneration but without evidence of morphologic aberrations.
The first historical account of MD was reported by Conte and Gioja in 1836.[1] They described two brothers with progressive weakness starting at age 10 years. These boys later developed generalized weakness and hypertrophy of multiple muscle groups, which are now known to be characteristic of the milder Becker MD. At the time, however, many thought that Conte and Gioja described tuberculosis; thus, they did not achieve recognition for their discovery.
In 1852, Meryon[2] reported in vivid details a family with four boys, all of whom were affected by significant muscle changes but had no central nervous system abnormality when examined at necropsy. Meryon subsequently wrote a comprehensive monograph on MD and even went on to suggest a sarcolemmal defect to be at the root of the disorder. He further suspected that the disorder is genetically transmitted through females and affects only males.
Guillaume Duchenne was a French neurologist who was already famous for his application of faradism (the use of electric currents to stimulate muscles and nerves) in the treatment of neurologic disorders when he wrote about his first case of MD.[3] In 1868, he gave a comprehensive account of 13 patients with the disease, which he called "paralysie musculaire pseudo-hypertrophique." Because Duchenne was already held in high esteem for his work in faradism and for his contributions to the understanding of muscle diseases, one of the most severe and classic forms of MD, Duchenne MD, now bears his name.
The advancement of molecular biology techniques illuminates the genetic basis underlying all MD: defects in the genetic code for dystrophin, a 427-kd skeletal muscle protein (Dp427). These defects result in the various manifestations commonly associated with MD, such as weakness and pseudohypertrophy. Dystrophin can also be found in cardiac smooth muscles and in the brain (accounting for the slight mental retardation associated with this disease).[4]
Minor variations notwithstanding, all types of MD have in common progressive muscle weakness that tends to occur in a proximal-to-distal direction, though there are some rare distal myopathies that cause predominantly distal weakness. The decreasing muscle strength in those who are affected may compromise the patient's ambulation potential and, eventually, cardiopulmonary function.
In addition, structural soft-tissue contractures and spinal deformities may develop from poor posturing caused by the progressive muscle weakness and imbalance, all of which can further compromise function and longevity. Equinovarus contractures start as flexible dynamic deformities and advance to rigid contractures. This altered anatomy prevents normal ambulation, proper shoe wear, and transfers (how patients can be picked up to transfer out of their chair).
Once wheelchair-bound, patients with MDs tend to develop worsening contractures and rapidly progressive scoliosis. On average, for each 10° of thoracic scoliosis curvature, the forced vital capacity (FVC) decreases by 4%.[5] In a patient with an already-weakened cardiopulmonary system, this decrease in FVC could rapidly become fatal.
The goal of orthopedic management is, therefore, to preserve or prolong patients' ambulatory status for as long as possible. This goal can be achieved with soft-tissue releases for contractures. If the patient develops significant scoliosis, which generally occurs after they stop walking, early stabilization of the spine should be considered.
NextMultiple proteins are involved in the complex interactions of the muscle membrane and extracellular environment. For sarcolemmal stability, dystrophin and the dystrophin-associated glycoproteins (DAGs) are important elements.[6, 7]
The dystrophin gene is located on the short arm of chromosome X near the p21 locus and codes for the large protein Dp427, which contains 3685 amino acids. Dystrophin accounts for only approximately 0.002% of the proteins in striated muscle, but it has obvious importance in the maintenance of the muscle's membrane integrity.[5]
Dystrophin aggregates as a homotetramer at the costomeres in skeletal muscles, as well as associates with actin at its N-terminus and the DAG complex at the C-terminus, forming a stable complex that interacts with laminin in the extracellular matrix. Lack of dystrophin leads to cellular instability at these links, with progressive leakage of intracellular components; this results in the high levels of creatine phosphokinase (CPK) noted on routine blood workup of patients with Duchenne MD.
Less-active forms of dystrophin may still function as a sarcolemmal anchor, but they may not be as effective a gateway regulator because they allow some leakage of intracellular substance. This is the classic Becker dystrophy. In both Duchenne and Becker MD, the muscle-cell unit gradually dies, and macrophages invade. Although the damage in MD is not reported to be immunologically mediated, class I human leukocyte antigens (HLAs) are found on the membrane of dystrophic muscles; this feature makes these muscles more susceptible to T-cell mediated attacks.
Selective monoclonal antibody hybridization was used to identify cytotoxic T cells as the invading macrophages; complement-activated membrane attack complexes have been identified in dystrophic muscles as well. Over time, the dead muscle shell is replaced by a fibrofatty infiltrate, which clinically appears as pseudohypertrophy of the muscle. The lack of functioning muscle units causes weakness and, eventually, contractures.
Other types of MDs are caused by alterations in the coding of one of the DAG complex proteins. The gene loci coding for each of the DAG complex proteins is located outside the X chromosomes. Gene defects in these protein products also lead to alterations in cellular permeability; however, because of the slightly different mechanism of action and because of the locations of these gene products within the body, there are other associated effects, such as those in ocular and limb-girdle type dystrophies (see the image below).
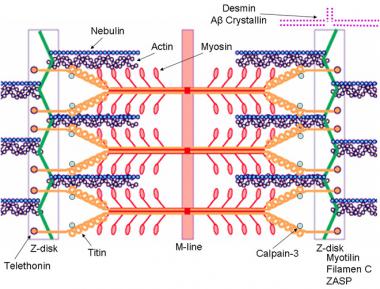 Schematic of the sarcomere with labeled molecular components that are known to cause limb-girdle muscular dystrophy or myofibrillar myopathy. Mutations in actin and nebulin cause the congenital myopathy nemaline rod myopathy, and the mutations in myosin cause familial hypertrophic cardiomyopathy. Image courtesy of Dr F. Schoeni-Affoher, University of Friberg, Switzerland.
Schematic of the sarcomere with labeled molecular components that are known to cause limb-girdle muscular dystrophy or myofibrillar myopathy. Mutations in actin and nebulin cause the congenital myopathy nemaline rod myopathy, and the mutations in myosin cause familial hypertrophic cardiomyopathy. Image courtesy of Dr F. Schoeni-Affoher, University of Friberg, Switzerland.
The etiology of MD is an abnormality in the genetic code for specific muscle proteins.[8] They all are classified according to the clinical phenotype, the pathology, and the mode of inheritance. The inheritance pattern includes the sex-linked, autosomal recessive, and autosomal dominant MDs. Within each group of heritable MDs (see below), several disorders exist. These are characterized by the clinical presentation and pathology.
Heritable MDs include the following:
In the X-linked forms of MD, such as the Duchenne and Becker dystrophies, the defect is located on the short arm of the X chromosome.[9] Hoffman and coworkers identified the locus of the defect in the Xp21 region, which includes approximately 2 million base pairs.[5] The gene codes for Dp427, which is a component of the cytoskeleton of the cell membrane.
Dystrophin is distributed not only in skeletal muscle but also in smooth and cardiac muscles and in the brain. The large size of the dystrophin gene explains the ease at which spontaneous new mutations can occur, as in Duchenne MD. The large size also allows mistakes in protein synthesis to occur at multiple sites.
Defects that interfere with the translation reading frame or with the promoter sequence that initiates synthesis of dystrophin lead to an unstable, ineffective protein, as in Duchenne MD. Disruption of the translation process further down the sequence leads to production of proteins of lower molecular weight that, although present, are less active and result in the milder variety of Becker MD.
Like Duchenne MD, Emery-Dreifuss MD is a sex-linked recessive disorder, but its defect is localized to the long arm of the X chromosome at the q28 locus.[10] Some authors, however, have cited case reports of similar findings in Emery-Dreifuss that were transmitted in an autosomal dominant pattern.[11] However, this finding is more of an aberration than a normal observation in Emery-Dreifuss MD.
In autosomal recessive conditions such as limb-girdle MD, the genetic defect is localized to the 13q12 locus.
In the autosomal dominant facioscapulohumeral MD, the defect is at the 4q35 locus. In distal MD, it is at the 2q12-14 loci.[12]
The incidence of MD varies, depending on the specific type of MD under consideration. Duchenne MD is the most common MD and is sex-linked, with an inheritance pattern of 1 case per 3500 live male births.[13, 14] One third of cases occurs as a result of spontaneous new mutations.[15] Becker MD is the second most common form, with an incidence of 1 case per 30,000 live male births.[16] Other types of MD are rare. For example, limb-girdle dystrophy occurs in only 1.3% of patients with MDs.
The incidence internationally is similar to that of the US for most of the dystrophies, except for the oculopharyngeal type, which is more common in French Canadians than in other groups.[17] Distal MD tends to occur in Sweden.
Despite modern advances in gene therapy and molecular biology, MD remains incurable. With proper care and attention, patients have a better quality of life than they would otherwise, but most still die by the time they are age 30 years, usually as a result of cardiopulmonary failure.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved