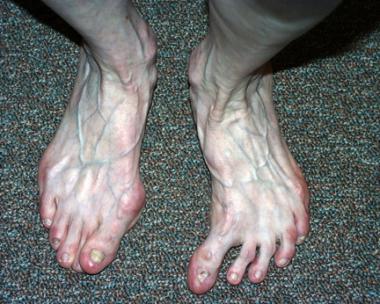
CHILD (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) syndrome is a rare disorder characterized by birth defects of several organ systems, including the skin, viscera, musculoskeletal system, and central nervous system. The earliest description of the syndrome has been attributed to Otto Sachs in 1903, who comprehensively described the clinical features of CHILD syndrome in an 8-year-old girl.[1] This was followed by a report in 1948 by Zellweger and Uelinger, who reported a patient with a "half-sided osteochondrodermatitis and nevus ichthyosiformis."[2] In 1980, Happle et al reviewed 18 previous cases and introduced two additional cases; they proposed the acronym CHILD syndrome for congenital hemidysplasia, ichthyosiform erythroderma, and limb defects.[3] In 2010, Knape et al reported the first case of CHILD syndrome with ocular manifestations in a patient with progressive bilateral optic nerve atrophy.[4]
Since then, other patients with a similar constellation of defects have been described under a number of designations, including unilateral ichthyosiform erythroderma, unilateral erythrokeratoderma, unilateral epidermal nevus, unilateral ectromelia, inflammatory variable epidermal nevus, and unilateral limb and skin deformities with congenital heart disease.
NextCHILD syndrome is inherited in an X-linked dominant fashion and involves a mutation in the NSDHL (NAD[P]H steroid dehydrogenase–like protein) gene.[5, 6, 7] The gene has been localized to Xq28 and encodes for 3beta-hydroxy sterol dehydrogenase, which catalyzes a step in the cholesterol biosynthetic pathway.[8] This enzyme is located both within the membranes of the endoplasmic reticulum and on the surface of intracellular lipid storage droplets. Several different types of mutations in the gene have been documented, including missense, nonsense, and stop mutations, all resulting in a loss of function of NSDHL.
Clinical variations in the extent of involvement are not thought to be secondary to the specific type of mutation, but rather the differences in the pattern of X inactivation.[9, 10] The striking laterality of the syndrome may arise from this impaired cholesterol processing, causing abnormal sonic hedgehog signaling, which, in embryogenesis, is important in spatial patterning.[11, 12]
United States
No precise data are available regarding the frequency of the disease; however, around 60 cases have been reported thus far in the literature.
Early death in persons with CHILD syndrome is most commonly due to cardiovascular malformations. However, central nervous system, skeletal, kidney, lung, and other visceral defects can contribute to significant morbidity.
The vast majority of reported cases occur in females because the disorder is X-linked dominant and lethal in males. However, 2 known cases have been reported in males, one with a normal 46,XY karyotype, which suggests an early postzygotic somatic mutation.[13]
CHILD syndrome is a congenital disorder. The dermatosis may be present at birth or may develop during the first few weeks of life and persists for the lifetime of the patient.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved