Hemangiomas are abnormal proliferations of blood vessels that may develop in any vascularized tissue. They may be asymptomatic or may cause symptoms such as pain and swelling. Considerable debate exists as to whether these lesions are neoplasms, hamartomas, or vascular malformations. Mulliken strongly supported classification of hemangiomas as neoplasms, whereas Godanich and Capanacci appeared to favor a hamartomatous classification.[1, 2]
There seems to be a consensus that the term hemangioma should refer to hemangiomas of infancy, which have a predictable natural history that includes absence at birth followed by a period of growth over 6-18 months and then a period of involution that may take several years. Hemangiomas affecting the musculoskeletal system are more accurately termed vascular malformations. These are present from birth and do not involute spontaneously.[3, 4]
Hemangiomas occur most often in skin or subcutaneous tissue, and dermatologists, pediatricians, and primary care medical physicians typically treat these readily identifiable processes. One common example is the senile or cherry hemangioma, which is a benign, self-limited, small, red-purple skin papule seen in elderly patients. Another is the strawberry nevus, which is seen in approximately 0.5% of infants and spontaneously involutes in the vast majority of cases. Visceral hemangiomas are far less common but may have greater consequences when they result in organ dysfunction.
Orthopedists most commonly are called upon to treat hemangiomas of the deep soft tissues and bone. Skeletal muscle is the most common site for hemangioma of the deep soft tissue. Intramuscular hemangiomas may cause symptoms such as pain and swelling for which patients seek treatment. Hemangioma of bone may be symptomatic or may be purely an incidental finding. Most commonly, hemangiomas are localized to a single area, but multiple hemangiomas may occur in a single individual in a process known as hemangiomatosis.[5, 6, 7, 8, 9]
Some authors have defined hemangiomatosis as multiple hemangiomas occurring in noncontiguous bones. Devaney et al defined skeletal-extraskeletal angiomatosis as a benign vascular proliferation involving the medullary cavity of bone and at least one other type of tissue.[10]
Rarely, hemangiomas may be associated with other pathologic processes, such as the consumptive coagulopathy of Kasabach-Merritt syndrome and tumor-induced osteomalacia. Gorham disease is a process of massive osteolysis, which is believed to be within the spectrum of hemangiomatous disease. Hemangiomas occurring in the setting of multiple enchondromatosis are part of the spectrum of Maffucci syndrome.
For patient education resources, see the Skin Conditions and Beauty Center, as well as Bruises.
NextHemangiomas are benign lesions with increased numbers of blood vessels. They can affect numerous tissue types (individually or in combination), including skin, subcutaneous tissue, viscera, muscle, synovium, and bone, but they do not spread to avascular tissue such as cartilage.
Gorham disease is a process in which variably progressive dissolution of bone occurs. This process may affect a single bone or may cross joint spaces. The etiology of this process is thought to be related to excess vascularity of the involved bone. The exact pathogenesis of this process is unknown at this time. Newer theories involve hormone and cytokine stimulation of osteoclast function, with heightened sensitivity of osteoclast precursors to interleukin-6 leading to increased bone resorption locally.[11]
Some have argued that epithelioid hemangiomas are not a distinct clinicopathologic entity but rather a misdiagnosed hemangioendothelioma, which is a tumor with malignant potential, unlike hemangioma.[12] Identification of WWTR1-CAMTA1 fusion as the genetic hallmark of epithelioid hemangioendothelioma regardless of anatomic location, provides an objective and powerful diagnostic tool that can be used to distinguish the two entities.[13] The WWTR1-CAMTA1 fusion was used in a retrospective study with good results to reinforce prior data that epithelioid hemangiomas are benign lesions and not low-grade malignant soft-tissue tumors.[14]
The etiology of hemangiomas is unclear. Angiogenesis likely plays a role in the vascular excess present. Cytokines, such as basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) are known to stimulate angiogenesis. Excesses of these angiogenic factors or decreases of angiogenesis inhibitors (eg, interferon gamma, tumor necrosis factor [TNF]-β, and transforming growth factor [TGF]-β) have been implicated in the development of hemangiomas.[15]
The etiology of Gorham disease is unknown but is thought to be related to increased vascularity of the affected bones. The resultant hyperemia has been hypothesized to uncouple the balance between osteoblasts and osteoclasts, leading to bone resorption at a far greater rate than bone formation.[16]
Although the etiology is not entirely clear, development of Kasabach-Merritt syndrome seems to be related to stagnation of blood flow within a large hemangioma, which leads to platelet trapping and a subsequent consumptive coagulopathy.
The mechanisms behind osteomalacia have not been fully elucidated.
Most deep soft tissue hemangiomas probably are asymptomatic and small and go completely unnoticed; therefore, the exact incidence and prevalence are impossible to determine with any degree of certainty. That said, intramuscular hemangiomas are uncommon compared with other types of hemangiomas.
Muscle hemangiomas accounted for 10 of 570 hemangiomas reported by Geschickter and Keasbey.[17] Watson and McCarthy estimated that intramuscular hemangiomas accounted for 0.8% of all benign vascular tumors.[18] Intramuscular hemangiomas occur most often in young people (range, 2 months to 66 years), with 80-90% presenting in persons younger than 30 years. Males and females are affected with nearly equal frequency.
Synovial hemangiomas are extremely rare. They can arise from any surface that is lined by synovium, particularly tendon or joint space. They typically occur in childhood and adolescence, with few reports of the disease in infants.[19]
Hemangiomas of bone accounted for approximately 1% of primary bone tumors from which biopsies were taken in Dahlin and Mirra's series.[20] Hemangiomas of bone may occur in patients of any age. Approximately 25% present in persons in the fifth decade of life; however, hemangiomas have been reported in patients as young as 2 years and as old as 77 years.
Approximately two thirds of osseous hemangiomas occur in the cranium or vertebrae, and hemangioma is the most common benign tumor of vertebrae. Vertebral hemangiomas are found in approximately 10-12% of autopsy specimens and have a slight predominance in females (2:1 ratio).[21]
Hemangiomatosis and skeletal-extraskeletal angiomatosis are rare conditions.
Gorham disease (ie, massive osteolysis, disappearing bone disease) is very rare (see the image below). The disease typically occurs in the second or third decade of life with most patients younger than 40 years.
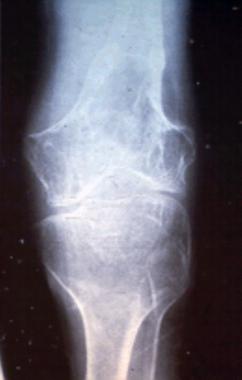 Radiograph of a patient with Gorham disease showing dissolution of bone.
Radiograph of a patient with Gorham disease showing dissolution of bone.
Kasabach-Merritt syndrome is a rare complication of large hemangiomas in which platelets are trapped and a consumptive coagulopathy ensues.
A significant number of intramuscular hemangiomas are associated with relatively mild symptoms, such as intermittent aching or discomfort with exercise. These may require no treatment and may have no significant sequelae. Unfortunately, those symptomatic enough to indicate treatment are those most likely to be incompletely excised, and thus to recur. Recurrence rates following surgery range from 18-50%.[22]
Outcomes for diffuse synovial hemangiomas are similar to those for intramuscular hemangiomas. Patients with localized synovial hemangiomas tend to have excellent results following surgical excision.
Many osseous hemangiomas remain asymptomatic, require no treatment, and have no significant sequelae.
In a small series, patients with symptomatic or locally aggressive vertebral hemangiomas treated with interventional or surgical modalities had good results, with either complete resolution of symptoms or only mild symptoms reported by the patients.[23] All patients returned to their prior levels of activity and reported minimal or no residual pain in daily activities.
Hemangiomatosis often becomes symptomatic during childhood, yet is nearly impossible to excise. Therefore, treatment with chemotherapy has been tried with variable success.
The rarity of Gorham disease precludes a clear assessment of its prognosis. Results of treatments have varied.
The natural history of many intramuscular hemangiomas is that of gradual fatty replacement, atrophy, and involution over time, as suggested by their greater frequency in individuals younger than 30 years and relative rarity in older adults. Many intramuscular hemangiomas are asymptomatic or produce only mild symptoms with activity, even during the active adolescent years. Treatment may be considered if pain is substantial, but because of the poor success rate of treatment and the apparent self-limited nature of most intramuscular hemangiomas, indications for treatment are few. The more localized the extent of the disease, the more likely it is to be controlled successfully by surgical excision.
The natural history of synovial hemangiomas may be similar to that of their intramuscular counterparts, but their rarity makes this difficult to document. The focal type more frequently is amenable to surgical excision than is the diffuse type.
Symptomatic or locally aggressive vertebral hemangiomas are usually treated conservatively when symptoms are limited to mild-to-moderate pain, whereas neurologic deficit and intractable pain are widely accepted in the literature as common indications for surgical intervention.[24] See Treatment for more details.
Both osseous hemangiomatosis and skeletal-extraskeletal angiomatosis often become symptomatic during childhood, with pain and diffuse swelling. Perhaps even more significantly, extraskeletal manifestations of hemangiomatosis can lead to hepatic dysfunction and cardiac complications. Because of the extensive nature of the disease, chemotherapy has been used with some success.
The natural history of Gorham disease is poorly defined. Extent and pace of bone loss are variable. No standard treatment is currently available for Gorham disease. Regression of lesions or stabilization of disease has been reported with steroids, radiation, surgery, bisphosphonates, zoledronic acid, and interferon-α.[11] Because of the unpredictable natural history and the potentially devastating effects of progressive disease, treatment generally should be instituted upon diagnosis.
Kasabach-Merritt syndrome is a potentially life-threatening coagulopathy that is related to platelet trapping in a large cavernous hemangioma. Approximately 30% of patients who develop this complication die from hemorrhage or infection. Surgical resection of the hemangioma often is difficult. Consequently, steroids, radiation therapy, interferon alfa-2a, and pentoxifylline have been used in attempts at treatment.
Tumor-induced osteomalacia results in diffuse osteopenia with marked hypophosphatemia, low serum calcium, and increased serum alkaline phosphatase. Because osteomalacia generally resolves with excision of the tumor, surgical treatment usually is indicated.
Contraindications to surgery are lack of symptoms, failure to attempt nonoperative measures, and threat to life or limb should surgery be performed.
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved