Dysplasia epiphysealis hemimelica (DEH), or Trevor disease, is a rare developmental disorder affecting the epiphyses in young children.[1] It is thought to be a variant of osteochondroma arising from an epiphysis.[2, 3, 4]
The first report of DEH in the literature was by Mouchet and Belot in 1926, who described the condition as a tarsal bone disorder and used the French term tarsomegalie.[5] In 1950, Trevor reviewed 10 cases of DEH and used the term tarsoepiphyseal aclasis.[6] In 1956, Fairbank reported 14 cases and coined the term dysplasia epiphysealis hemimelica.[7, 8, 9, 10, 11, 12, 13, 14, 15, 16]
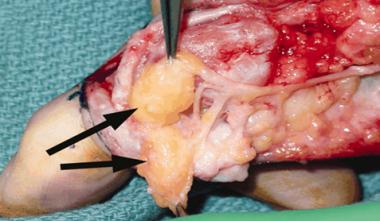
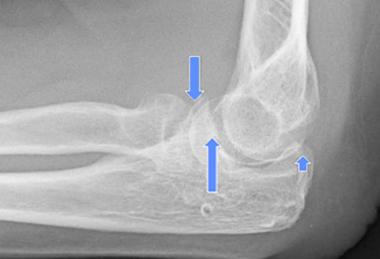
The typical DEH patient presents during childhood or adolescence. The characteristic lesions are intra-articular, are hemimelic (involving only half of the joint), have a predilection for the lower extremity, and may be single or multiple.[1] The differential diagnoses include chondroblastoma, osteochondroma, and enchondroma.[9, 2, 3, 17] Most cases are treated surgically.
Next
Many theories exist regarding the pathophysiology of DEH. Connor et al suggested that the fundamental defect was an abnormality of the regulation of cartilage proliferation in the affected epiphysis, resulting in cartilaginous exostosis.[18]
Trevor considered DEH to be a congenital error in epiphyseal development that affects the limb buds during early fetal life; it was thought to involve an altered process of cell proliferation at the superficial zone of articular cartilage, allowing for persistent proliferation and production of a large cartilaginous mass.[6]
Fairbank suggested that the disorder derived from a localized disturbance of the preaxial or postaxial apical cap of the limb bud in early fetal development.[7, 9]
The etiology of DEH is unknown; the disease does not appear to be genetically transmitted.
The incidence of DEH has been estimated to be 1 case per million population. It is a rare disorder both in the United States and internationally. Although DEH may occur at any age, it usually manifests in childhood and early adolescence. DEH is more commonly found in males, with a male-to-female ratio of 3:1. No racial predilection is known to exist.
DEH is a benign disorder, and no cases of malignant transformation have been reported. Its natural history is that of a lesion that continuously increases in size until skeletal maturity; therefore, the long-term prognosis for untreated lesions involving the weight-bearing surface of the joint, though unreported, is one of a progression toward pain and arthrosis.
DEH, though an uncommon condition, can result in considerable disability as consequence of the direct involvement of the articular surface of the joint. Treatment with surgical excision offers the best results, but corrective osteotomy and reoperation for recurrent lesion may also be required.[19, 20]
Clinical Presentation
Copyright © www.orthopaedics.win Bone Health All Rights Reserved