Primary spinal tumors fall into a distinct category because their timely diagnosis and the immediate institution of treatment have an enormous impact on the patient's overall prognosis and hope for a cure.
Generally, with spinal pathology, problems that arise are either chronic problems related to degenerative disease or deformity or acute manifestations of traumatic sequelae. When considering tumors of the spine, one must consider the different tissue types around the spinal column. The presence of neural tissue, meningeal tissue, bone, and cartilage make any of these tissue types a possible nidus for neoplastic change. Also, metastatic lesions may spread to the spine from distant primary tumor sites by hematogenous or lymphatic routes.
Primary nonlymphoproliferative tumors of the spine are uncommon and make up fewer than 5% of bone neoplasms, accounting for fewer than 2.5-8.5 primary spine tumors per 100,000 people per year. Metastatic disease of the spine is much more common. Approximately 40-80% of patients who die of cancer have bony metastases at the time of death, with the spine being the most common metastatic skeletal location.
Neoplastic disease, however, can present with back pain that is indistinguishable from back pain resulting from more benign causes. Therefore, the physician caring for patients complaining of back pain is faced with the challenge of distinguishing benign causes from those that can be neurologically or systemically devastating and prescribing the appropriate treatment.
This distinction sometimes can be difficult to make because of the complicated architecture of the spine. The physician must consider differential diagnoses of degenerative processes, infections, muscular strains, neurologic impingements, and, finally, neoplastic processes. With thorough history taking, physical examination, and diagnostic imaging, the physician can acquire enough information to efficiently make the correct diagnosis.[1]
NextThe most common clinical presentation associated with all spine tumors is back pain that causes the patient to seek medical attention. Back pain is the most frequent symptom for patients with either benign or malignant neoplasms of the spine. Neurologic deficits secondary to compression of the spinal cord or nerve roots also can be part of the presentation.
The degree of neurologic compromise can vary from slight weakness or an abnormal reflex to complete paraplegia, depending on the degree of encroachment. The loss of bowel or bladder continence can occur from neurologic compression or can be secondary to a local mass effect from a tumor in the sacrococcygeal region of the spine, as occurs in chordomas. Systemic or constitutional symptoms tend to be more common with malignant or metastatic disease than in benign lesions.
For these patients, workup should include a complete blood count and differential, a basic serum chemistry profile, erythrocyte sedimentation rate, or C-reactive protein to help distinguish between neoplastic and infectious processes. Elevations in serum calcium or alkaline phosphatase also can provide evidence for neoplastic bone processes. Specific studies, such as serum electrophoresis or urine electrophoresis, also can be performed to evaluate the likelihood of multiple myeloma or plasmacytoma.
Imaging studies for the workup of spine tumors include plain radiography, computed tomography (CT), magnetic resonance imaging (MRI), and technetium bone scanning.[2, 3, 4]
The first-line imaging study should be plain radiography to evaluate the trabecular architecture of the spine. Anteroposterior (AP), lateral, and oblique views may be required. These studies should be evaluated with respect to both what the tumor is doing to the bone and, conversely, what the bone is doing to the tumor. The blastic or lytic nature of the lesion should be noted. The general location of the lesion within the bone, the integrity of the cortex, and the presence of fractures or soft tissue masses are important findings (see the images below).
 Spinal tumors. Coned-down view of hemangioma in the thoracic spine.
Spinal tumors. Coned-down view of hemangioma in the thoracic spine.
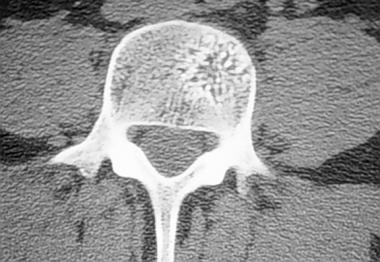 Spinal tumors. Axial CT scan of hemangioma in a lumbar vertebra.
Spinal tumors. Axial CT scan of hemangioma in a lumbar vertebra.
The ultimate way to make the diagnosis and ascertain the specific tumor type is by performing a biopsy of the spine lesion after all radiographic studies have been completed. Biopsies can be performed with open or by percutaneous image-guided technique. Percutaneous needle biopsies may not supply adequate tissue for the diagnosis of a primary tumor of the spine.
The basic principles of biopsy technique also apply to tumors of the spine. The surgeon performing the biopsy should take the most direct route to the tumor, with the least potential to contaminate adjacent compartments. The biopsy tract should be placed in line with the future incision site for surgical resection of the tumor, so that the biopsy tract can be excised with the specimen en bloc.
Meticulous hemostasis must be obtained, and a drain must be placed to prevent hematoma formation, which can dissect the soft tissue planes and contaminate adjacent compartments. The drain should exit the skin in line with the incision so that it, too, can be excised with the final specimen.
The histologic findings vary according to the tumor types as described above. The following list revisits the primary tissue type associated with some of the tumors of the spine:
Bone-producing tumors of the spine include the following (see the images below):
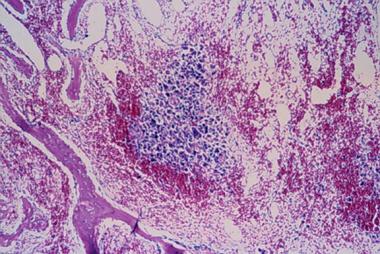 Spinal tumors. Aneurysmal bone cyst histology.
Spinal tumors. Aneurysmal bone cyst histology.
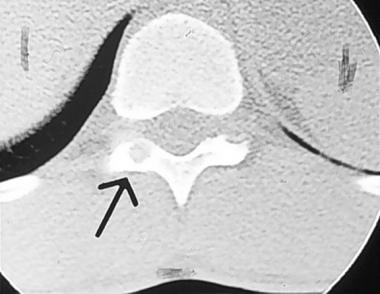 Spinal tumors. Axial CT scan of thoracic vertebra, which demonstrates the nidus of the osteoid osteoma in the posterior elements.
Spinal tumors. Axial CT scan of thoracic vertebra, which demonstrates the nidus of the osteoid osteoma in the posterior elements.
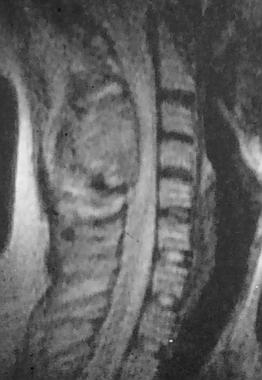 Spinal tumors. MRI of the osteoblastoma in the posterior elements of C3 and C4 seen on the previous x-ray image.
Spinal tumors. Histology of osteoblastoma at low magnification.
Spinal tumors. MRI of the osteoblastoma in the posterior elements of C3 and C4 seen on the previous x-ray image.
Spinal tumors. Histology of osteoblastoma at low magnification.
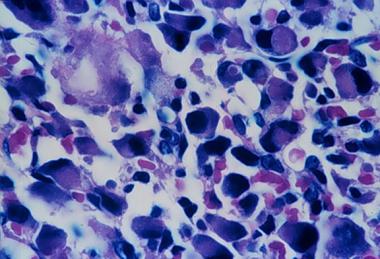 Spinal tumors. Higher magnification of osteoblastoma.
Spinal tumors. Higher magnification of osteoblastoma.
Cartilage-producing tumors of the spine include the following:
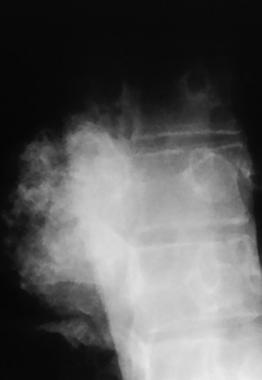 Spinal tumors. Coned-down view of lateral thoracic spine in a patient with a chondrosarcoma.
Spinal tumors. Coned-down view of lateral thoracic spine in a patient with a chondrosarcoma.
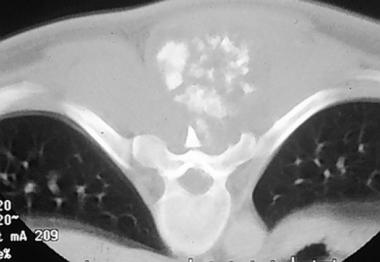 Spinal tumors. Axial CT scan at the level of the chondrosarcoma seen on the previous x-ray image.
Spinal tumors. Axial CT scan at the level of the chondrosarcoma seen on the previous x-ray image.
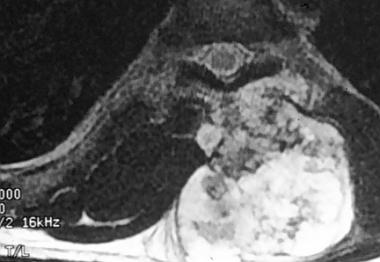 Spinal tumors. T2-weighted MRI scan of the chondrosarcoma in the same patient.
Spinal tumors. T2-weighted MRI scan of the chondrosarcoma in the same patient.
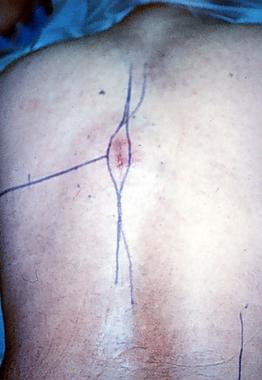 Spinal tumors. Photograph of the patient's back at the time of surgery exhibiting the course of the definitive incision to excise the chondrosarcoma en bloc with the previous biopsy tract included with the resection.
Spinal tumors. Photograph of the patient's back at the time of surgery exhibiting the course of the definitive incision to excise the chondrosarcoma en bloc with the previous biopsy tract included with the resection.
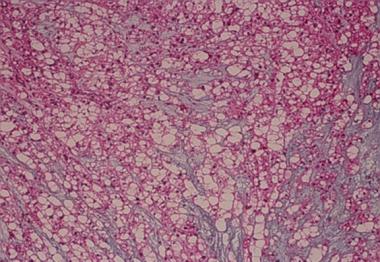
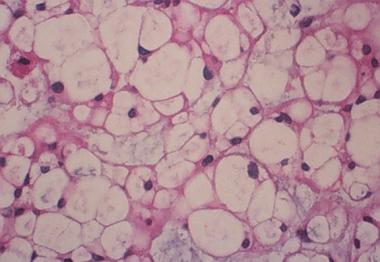
 Spinal tumors. Histology of the chondrosarcoma at 40 times magnification.
Spinal tumors. Histology of the chondrosarcoma at 40 times magnification.
Lymphoproliferative tumors include the following:
Chordoma is a tumor of notochordal origin that may be identified by the characteristic physaliferous cells (see the images below)
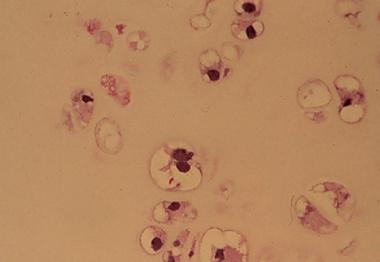
 Spinal tumors. Chordoma histology.
Spinal tumors. Chordoma histology.
 Spinal tumors. Higher magnification of chordoma histology demonstrates the characteristic physaliferous cells.
Spinal tumors. Higher magnification of chordoma histology demonstrates the characteristic physaliferous cells.
Ewing sarcoma is a malignant round cell tumor of childhood that is associated with large sheets of homogenous small, round, blue cells.
The spine consists of 33 vertebrae that form the bony spinal column. The spinal column can be divided into the cervical, thoracic, lumbar, and sacrococcygeal regions. Although morphologically distinct, each vertebra in the subaxial cervical, thoracic, and lumbar spine has a complex architecture, consisting of a vertebral body, pedicles, laminae, and spinous and transverse processes.
The bony canal provides protection and support to the fragile spinal cord and nerve roots within the dural sac. The soft tissues surrounding the bony spine vary by location from the thick dorsal paraspinous musculature to the vital organs and vessels within the mediastinal, thoracic, peritoneal, and retroperitoneal spaces.
The relevant anatomy discussed above is frequently the limiting factor in determining contraindications for surgical excision of spine tumors. The morbidity of the tumor, the tumor's malignant potential, and the patient's overall prognosis must be compared to the morbidity and potential mortality of radical resection of a tumor near the spinal cord, the aorta, or the heart.
The degree of associated blood loss and the overall health of the patient also must be taken into consideration in considering a resection. If the patient is known to have metastatic or systemic tumor involvement, this may be a contraindication to radical resection of a paraspinous tumor, which may render the patient paralyzed.
Weinstein and McLain,[5] and Boriani et al[6] developed a descriptive staging system for spine tumors based on the principles of the Enneking staging system for primary bone tumors of the extremities. In this staging system, the transverse extension of the vertebral tumor is described with reference to 12 radiating zones (numbered 1-12 in clockwise order) and five concentric layers (A to E) from the paravertebral extraosseous compartments to the dural involvement. The longitudinal extent of the tumor is recorded according to the levels involved.
Based on an understanding of the biologic behavior of the tumor, the oncologic staging aids the surgeon to decide what surgical margin provides the best chance for complete tumor resection and possible cure. This system is complex and sometimes difficult to apply clinically.
The Enneking classification of benign lesions applies to benign spine tumors, as follows:
Stage 1 lesions are usually asymptomatic and are discovered incidentally. Stage 2 lesions usually present with symptoms; most commonly, pain is in the area of the lesion. Stage 3 lesions are locally aggressive and can actually metastasize.[7]
Also termed a bone island, enostosis is a mass of calcified medullary defects of lamellar compact bone with haversian systems found within the cancellous portion of the bone. It occurs most frequently in the thoracic and lumbar spine, usually between T1 and T7 and between L1 and L2. Enostosis is one of the most common lesions to involve the spine.
Enostoses are usually stage 1 lesions and are discovered incidentally. Most remain stable, but some may slowly increase in size. Resnik et al determined the incidence of enostosis to be approximately 14% in cadavers.[8]
Radiographically, enostoses are circular or oblong osteoblastic lesions with a spiculated margin, which gives it the appearance of thorny periphery. An abrupt transition from normal to the sclerotic bone is exhibited on the x-ray. Bone scan findings are usually normal, and magnetic resonance imaging (MRI) demonstrates low signal intensity with normal surrounding intensity.
Enostosis sometimes can be confused with osteoblastic metastatic disease. Enostosis can be differentiated by lack of activity on bone scan, by the normal appearance of adjacent bone, by its thorny margins, and by lack of a primary tumor for metastasis. If the enostosis exhibits an increase in diameter of greater than 25% in 6 months, a biopsy should be performed.
Osteoid osteomas usually present in children aged 10-20 years, with a male predominance. They involve the axial skeleton only 10% of the time. In the spine, 59% of osteoid osteomas are found in the lumbar region, 27% in the cervical region, 12% in the thoracic region, and 2% in the sacral region.[9, 10]
Osteoid osteomas are usually stage 2 lesions and are actively symptomatic. They can result in painful scoliosis, radicular pain, gait disturbances secondary to pain and splinting, and muscular atrophy. Symptoms usually are relieved or ameliorated by nonsteroidal anti-inflammatory drugs (NSAIDs) or salicylates. In the spine, osteoid osteomas occur 75% of the time in the posterior elements, either the pedicles, facets, or laminae. Osteoid osteomas occur 7% of the time in the vertebral body and 18% of the time in the transverse and spinous processes.
On plain x-ray, osteoid osteomas appear as a round or oval radiolucent nidus, with a surrounding rim of sclerotic bone. An area of central calcification may be present, but this classic appearance may be obscured by complex spinal architecture. Bone scan shows marked increased uptake by the nidus, and a double intensity pattern may exist. Computed tomography (CT) is the criterion standard for radiographic diagnosis. The nidus is a well-defined area of low attenuation with or without central calcification surrounded by an area of sclerosis.
The nidus is usually smaller than 1.5-2.0 cm, composed of microscopic well-organized trabecular bone with vascular fibrous connective tissue stroma surrounded by reactive cortical bone.
Treatment is accomplished by resection of the nidus via an open surgical approach or by percutaneous CT scan-guided resection. Percutaneous radiofrequency ablation of the nidus has been performed with acceptable results.[11]
Histologically similar in appearance to osteoid osteoma, osteoblastoma is behaviorally very different. Demographically, it occurs in young patients in the second or third decade of life. A 2:1 male-to-female predominance exists. The lesion is distributed equally in the cervical, thoracic, and lumbar segments of the spine. The posterior elements are involved in 55% of cases, but the tumor can extend to the vertebral body in 42% of cases.
Patients typically complain of dull localized pain and paresthesias, paraparesis and, if the tumor is large enough and encroaching on the spinal cord, paralysis.
Osteoblastomas are expansile lesions with multiple small calcifications and a peripheral scalloped and sclerotic rim. In more aggressive lesions, osseous expansion, bone destruction, infiltration of the surrounding tissue, and intermixed matrix calcification are present. Some 50% of osteoblastomas are radiolucent, and 20% are osteoblastic.
Marked radionucleotide uptake is exhibited on bone scan. CT scan demonstrates areas of mineralization, expansile bone remodeling, and sclerosis or a thin osseous shell at its margins. MRI is nonspecific but is the criterion standard to assess the effect of the tumor on the cord and surrounding tissues.
Osteoblastomas are typically larger than 2.0 cm in diameter with histologic features of interconnecting trabecular bone and fibrovascular stroma similar to, but not as well organized as, osteoid osteoma. They can have an aneurysmal bone cyst component in 10-15% of cases.
Wide local resection is the treatment of choice whenever possible. This sometimes is limited by the proximity of vital vessels or neural tissue in the spine. A 10-20% recurrence rate exists for conventional osteoblastomas. Aggressive osteoblastomas have a recurrence rate of approximately 50% if wide margins are not attained. These tumors are not radiosensitive.
Aneurysmal bone cysts (ABCs) typically affect young patients, with 80% occurring in people younger than 20 years. The spine is involved 12-30% of the time. The thoracic spine is affected most commonly, followed by the lumbar and cervical spines. Sacral involvement is rare.[12]
ABCs of the spine usually present as expansile areas of bone remodeling in the posterior elements. Extension into the vertebral body can occur 75% of the time. The lesion may have a thin outer periosteal rim of bone, and septations within the mass may be apparent. The mass may extend into adjacent vertebrae, discs, ribs, and paravertebral soft tissues.
The bone scan exhibits peripheral increased uptake with a central "cold area" creating a donut sign. If angiography is performed, the mass is found to be hypervascular 75% of the time. CT scans and MRIs are used to confirm the cystic nature of the lesion as well as the tumor extension into surrounding tissues and the tumor's relationship to the spinal canal. Single or multiple fluid/fluid levels sometimes can be visualized on MRI. MRIs with gadolinium demonstrate enhancement of the periosteal rim and septations and not the cystic spaces.
ABCs are characteristically multiloculated blood-filled spaces that are not lined by endothelium. They are not vascular channels. Primary ABCs are believed to result from microtrauma to the bone with local circulatory disturbance. Other underlying neoplasms, such as giant cell tumors (GCTs), osteoblastomas, chondroblastomas, or osteosarcomas, produce secondary ABCs. These other neoplasms produce venous obstruction and possible arteriovenous malformations and set the stage for ABC formation. Most ABCs are considered primary (65-95%).
Because of the locally aggressive behavior of spinal ABCs, their treatment can be problematic. The severe morbidity that can be associated with complete resection is caused generally by danger to surrounding vascular or neural elements. ABCs can have a recurrence rate of 20-30% or higher, depending on the degree of resection. Preoperative embolization therapy and radiation may help shrink the tumor's size and decrease the amount of intraoperative blood loss associated with resection.
Osteochondromas make up 4% of all solitary spine tumors.[13] They also are commonly referred to as exostosis. Spinal lesions are encountered in 7-9% of patients with multiple hereditary exostoses (MHE). Osteochondromas occur in patients aged 20-30 years. Patients with MHE tend to develop the osteochondroma at a younger age; they also tend to experience neurologic deficits and myelopathy more frequently (77% of the time) than the patient with solitary osteochondroma (34%). A male predominance exists.
Osteochondromas are more common in the cervical spine, especially at C2. The posterior elements usually are involved. The lesions are believed to arise secondary to trapping of the physeal cartilage outside the growth plate during skeletal development.
Making the diagnosis of osteochondroma in the spine on plain radiography can be difficult unless the lesion is large and protruding posteriorly from a spinous process. In fact, 15% of patients with osteochondromas of the spine have normal appearing x-rays. CT scan is the study of choice to detect the exostosis and determine its relationship to the surrounding soft tissue and spinal canal.
T1-weighted MRI scans reveal a central area of high signal intensity, which represents yellow marrow. This area has intermediate intensity on T2-weighted images. The cortex of the exostosis has low signal intensity. The hyaline cartilage cap of the exostosis is best evaluated with MRI and appears with low signal intensity on T1 and high intensity on T2. The cartilage cap should be less than 2 cm in adults. Lesions with cartilage caps greater than 2 cm should be suspected of malignant transformation to chondrosarcoma.
Qualitatively, the bone composing an osteochondroma is normal. Abnormal bone growth occurs at and as a result of the cartilage cap. A continuity of the lesion with the marrow and cortex of the underlying bone is present. The exostosis may be sessile or pedunculated.
Complete surgical resection is usually curative. Clinical symptoms improve in 89% of patients following removal of the exostosis. Incomplete resection can lead to recurrence of the lesion.
Giant cell tumors of the spine account for only 7% of all GCTs in the body. The spine is the fourth most common site for the occurrence of GCTs. Most spinal GCTs occur in the sacrum, followed by the thoracic, cervical, and lumbar regions. GCTs are more common in women and occur in the third to fifth decades of life. They can increase dramatically in size during pregnancy secondary to hormonal influences. Symptoms include pain with radicular pattern. With neurologic impingement, weakness and sensory deficits also can be manifest.
Spinal GCTs are usually radiolucent and expansile lesions. They do not exhibit active matrix production. When present in the sacrum, these lesions are large with destruction of the sacral foraminal lines on plain x-rays. GCTs usually can involve both sides of the midline and can extend past the sacroiliac joints bilaterally. When present in sites proximal to the sacrum, they usually are found in the vertebral body.
The classic findings of GCT on technetium bone scan include diffuse radionucleotide uptake with areas of central photopenia and increased peripheral uptake. Angiography illustrates that most GCTs are hypervascular lesions. CT scans demonstrate soft tissue attenuation with well-defined margins and a thin rim of sclerotic bone. MRI exhibits characteristic heterogeneous signal intensity with low-to-intermediate intensity on both T1- and T2-weighted images.
Most GCTs are benign; malignant GCTs occur in only 5% of cases. Malignant GCTs usually are related to previous irradiation in the vicinity of the tumor. Although most GCTs are benign, the lesions are locally aggressive, and their size and location may not allow complete resection. Those that cannot be excised en bloc should be curetted. Radiation is reserved for surgically inaccessible tumors. Selective arterial embolization also can be used in the management of these tumors. Recurrence rates can be as high as 40-60%.
Levine and Crandall offer a summary of the treatment of primary malignant tumors of the spine.[14] For treatment of astrocytomas, a less common spinal tumor, see Minnehan et al.[15] For treatment for nonambulatory patients, see Kondo et al.[16] Palliative surgery for metastatic thoracic and lumbar tumors is presented by Cho and Sung.[17]
Kaloostian et al conducted a review of the literature regarding treatment and outcomes of patients with metastatic disease or primary tumors of the spinal column.[18] They reported that en-bloc resection is the mainstay of treatment for malignant primary tumors of the spinal column, whereas intralesional resection is generally appropriate for benign primary tumors. Low-quality evidence supports the use of chemotherapy in select primary tumors. Radiation therapy is often used for incompletely resected or unresectable lesions.
According to this review, surgical considerations for the treatment of metastatic disease (see Metastatic Tumors in the Spine) are more nuanced and require consideration of patient performance status and the pathology of the primary tumor.[18] Treatment of metastatic and primary tumors of the spinal column requires a multidisciplinary approach.
Chondrosarcoma is the second most common nonlymphoproliferative tumor of the spine. Chondrosarcomas make up 7-12% of all spine tumors, and the spine is the primary site in 3-12% of all chondrosarcomas. Men are affected two to four times more frequently than women. The mean age of presentation is 45 years. The thoracic spine is the most common site, but chondrosarcomas can occur at all levels of the spine. The most common symptoms are pain, a palpable mass, and neurologic complaints in 45% of patients.
Plain radiographs of chondrosarcomas typically demonstrate bone destruction. The lesions may be apparent in the vertebral body 15% of the time, in the posterior elements 40% of the time, or in both 45% of the time. In 70% of patients, the characteristic chondroid matrix in the form of rings and arcs is apparent on x-ray. Cortical destruction with soft tissue extension is best observed on CT scan or MRI.
Chondrosarcomas that arise from malignant transformation of osteochondromas are observed as a thickening of the cartilaginous cap. Involvement of the adjacent vertebral levels by extension through the disc is observed in 35% of all lesions. On CT scan or MRI, mineralization is usually apparent in the soft tissue component of the lesion. The radionucleotide uptake by the lesion is intense and has a heterogeneous appearance on bone scan.
Chondrosarcomas are relatively low-grade lesions (grade I or II). Most lesions are primary chondrosarcomas rather than secondary chondrosarcomas that arise from the malignant degeneration of osteochondromas, as previously noted. Chondrosarcomas have relatively sparse cartilaginous stroma with a surrounding pseudocapsule. Examination under higher magnification reveals atypical nuclei with several mitotic figures per high-powered field.
Surgical resection by vertebral corpectomy and strut bone grafting sometimes may be necessary for complete excision. Cure is possible when complete resection can be achieved; this is possible 25% of the time. If wide marginal resection cannot be achieved, the tumor recurrence results in death in 74% of cases. The mean survival for all patients with chondrosarcomas is 5.9 years according to Shives et al.[19]
Adjunctive treatment with radiation is controversial for these tumors. Chemotherapy is used sometimes to help decrease the size of the mass with high-grade chondrosarcomas and dedifferentiated chondrosarcomas. Metastases of chondrosarcoma depend on the grade of the primary chondrosarcoma. The lungs are the most frequent sites of metastasis.
Ewing sarcoma is the most common nonlymphoproliferative primary malignant tumor of the spine in children. Lesions of the spine make up 3-10% of all primary sites of Ewing sarcoma. Metastatic foci of Ewing sarcoma involving the spine are more common than primary lesions of the spine. Patients with Ewing sarcoma usually present when aged 10-20 years.
The most common site of occurrence in the spine is the sacrococcygeal region followed by the lumbar and thoracic segments. Ewing sarcoma rarely occurs in the cervical spine. Lesions are centered primarily in the vertebral body but they can extend into the posterior elements.[20]
Plain x-rays reveal permeative bone lysis, osseous expansion, or sclerosis. Diffuse sclerosis is observed in 69% of spinal lesions and is associated with osteonecrosis. CT and MRI demonstrate osseous involvement as well as surrounding soft tissue involvement. However, MRI is nonspecific.
Tissue from a Ewing sarcoma is composed of sheets of small, round, blue cells divided by septa, scant cytoplasm, and abundant collagen. Areas of osteonecrosis are found in spinal lesions. These correspond to the sclerotic areas observed on plain x-rays, as discussed above. Genetically, patients with Ewing sarcoma are found to have an 11;12 chromosomal translocation.
Before the advent of chemotherapy, the survival rate for patients with Ewing sarcoma was dismal because of the inability to completely resect these lesions, especially in the axial skeleton.
Radiation and chemotherapy are the current mainstays of treatment of Ewing sarcoma in the spine, achieving almost 100% local control with an 86% long-term survival rate in patients with spinal Ewing nonsacral sarcomas. Sacral tumors have a 62% local control rate and only 25% long-term survival rate because of the tendency for delayed clinical presentation and larger tumor size. The most important prognostic indicator for survival of Ewing sarcoma is the tumor's response to chemotherapy.
Osteosarcomas of the spine are rare, making up only 0.6-3.2% of all osteosarcomas and only 5% of all primary malignant tumors of the spine. They typically present in patients in the fourth decade of life and have a male predominance. Osteosarcomas are found at all levels of the spine but are most common in the lumbosacral segments. Eccentric involvement of the vertebral body with extension into the posterior elements is common.
Patients often present with pain and a palpable mass. Neurologic symptoms, ranging from sensory deficits to paresis, are found in 70-80% of patients. Serum alkaline phosphatase may be elevated.
Plain x-rays of spinal osteosarcomas reveal a densely mineralized matrix, giving rise to the term ivory vertebrae. A loss of vertebral height often occurs, with sparing of the adjacent disc. Purely lytic lesions also have been described. CT and MRI are useful to evaluate the extent of bony and soft tissue involvement. If a large amount of mineralized matrix is present, the lesion may appear with low signal intensity on all MRI sequences.
Most osteosarcomas are blastic lesions (osteoblastic, chondroblastic, or fibroblastic). Osteosarcomas can arise primarily or secondarily from an exposure to radiation. Secondary osteosarcomas can have a latency of up to 20 years. Spinal osteosarcomas also have been found in patients with Paget disease.
Surgical resection is the rule; however, resection of spine lesions is often incomplete due to the size and location of the tumor at the time of presentation. Adjuvant chemotherapy and radiation therapy often are employed with varying degrees of utility. Spinal osteosarcomas have a dismal prognosis, with deaths usually occurring within the first year of diagnosis. Only a few patients have been reported to survive longer than 2 years.
Luschka first described chordoma morphologically in 1856 in Virchow's lab. The discovery of the notochordal nature of the tumor and the coining of the term chordoma is credited to Ribbert in 1894.
Chordomas are uncommon, accounting for 2-4% of all primary malignant bone tumors with a prevalence of 0.51 per million. However, they are excluding lymphoproliferative tumors and metastases, the most common primary malignant tumor of the spine in the adult.
As Ribbert described, chordomas arise for the notochord remnant. The notochord normally evolves into the nucleus pulposus of the intervertebral discs. Nonneoplastic notochord vestiges also are found at the midline of the sphenooccipital synchondrosis and in the sacrococcygeal regions. The locations in which chordomas occur parallel these vestigial distributions.
Regarding chordoma prevalence, 30-35% occur in the sphenooccipital region, 50% in the sacrococcygeal region (especially S4-S5), and 15% occur in the other spinal segments.
Interestingly, chordomas have not been reported to arise from the intervertebral discs. Chordomas occur most commonly in patients aged 30-70 years, with a peak incidence in the fifth to sixth decades of life. Sphenooccipital lesions have equal sex distributions but sacrococcygeal lesions have a 3:1 male-to-female ratio.
Presentation of chordomas is often subtle, with a gradual onset of pain, numbness, motor weakness, and constipation or incontinence. Constipation is a uniform finding in most patients with sacrococcygeal lesions. Chordomas are typically slow growing lesions and are often very large at the time of presentation.
On plain x-ray, chordomas appear as a destructive lesion of a vertebral body in the midline, with a large associated soft tissue mass. In sacrococcygeal lesions, osseous expansion is frequent and may extend across the sacroiliac joints. Mineralization within the tumor may be observed on the plain x-rays of 50-70% of sacrococcygeal lesions. The mineralization is amorphous and predominates in the periphery of the lesion.
Lesions in spinal segments above the sacrum are less expansile and demonstrate evidence of calcification in only 30% of cases. They may have areas of sclerosis in 43-62% of cases. The intervertebral discs above or below a chordoma may be involved and narrowed in a manner that simulates infection. The lesion can make its way through the intervertebral disc to infiltrate an adjacent level. This occurs in approximately 11-14% of cases.
CT demonstrates both the osseous and soft tissue components of the tumor. Coronal and sagittal reconstructions of the CT scan are helpful in assessing neural foraminal and sacroiliac joint involvement. MRI scans are an important adjunct in the workup of chordomas. The lesions appear with low-to-intermediate signal intensity on T1 images with very high signal intensity on T2 images, reflecting the high water content of chordomas. Enhancement occurs following intravenous contrast on both CT and MRI.
Chordomas are lobulated neoplasms, which usually are contained within a pseudocapsule. Histology of these lesions reveals long cords of physaliphorous cells. Physaliphorous cells are clear cells containing intracytoplasmic vacuoles with abundant intracellular and extracellular mucin. Sarcomatous chondroid, osteoid, or fibroid elements may be found within the chordoma.
Surgical resection is the rule. Adjuvant postoperative radiation therapy, proton beam therapy, and brachytherapy all have been used with varying results. The prognosis depends on whether the tumor can be resected completely. The location of the lesion and the size at presentation often necessitate incomplete resection. The treatment of sacral chordoma is an arduous clinical undertaking that requires a multidisciplinary approach and attention to detail from the outset.
Despite aggressive, well-planned surgical management and adherence to strict surveillance protocols, frequent recurrence and the late onset of metastatic disease are to be expected in a substantial proportion of patients, especially those with a large chordoma or one at a more cephalad level. Adequate surgical treatment results in substantial functional impairment and numerous complications; however, it does offer the possibility of long-term disease-free survival.[21]
Persons with sacrococcygeal tumors often have improved survival because the surrounding structures are relatively more expendable and allow a more complete resection. Persons with sacrococcygeal lesions typically have 8-10 years survival, as opposed to 4-5 years survival for persons with chordomas in other spinal sites. Death usually is related to local recurrence and invasion rather than metastatic disease. Chordomas can metastasize. The most common sites of metastases are the liver, lungs, regional lymph nodes, peritoneum, skin, and heart.
Multiple myeloma is a systemic disease that affects middle-aged people and is characterized by areas of local bone destruction. Multiple myeloma is the most common primary malignancy of bone and the spine. The underlying cell line is the malignant plasma cell, which produces abnormal quantities of immunoglobulins.
The presentation of patients with myeloma is similar to that of other spine tumor patients. Patients complain of pain that may be worse at night. The laboratory workup for these patients should include a complete blood count with differential looking for anemia and thrombocytopenia, an elevation of the erythrocyte sedimentation rate, and a decrease in the serum albumin with increased total serum protein. The abnormal production of immunoglobulins can be detected on serum or urine electrophoresis and can be used to confirm the diagnosis.
Radiographically, skeletal survey is used to screen for lesions that can occur throughout the skeleton. Bone scans have a high false-negative rate and are not optimal studies for the evaluation of myeloma. Once a lesion is detected in the spine, CT, MRI, or both should be performed to assess the destruction of the vertebrae and the effect of this destruction on the surrounding neurologic and paraspinous tissues.
Multiple myelomas are generally sensitive to radiation therapy and chemotherapy. Surgery for stabilization is indicated in myelomas of the spine when destruction of the vertebral body exists to such an extent that collapse and possible kyphosis with canal compromise could result.
Prophylactic posterior stabilization can be carried out with segmental instrumentation in cases prior to fracture. Anterior strut grafting or cage reconstruction may be necessary once fracture and collapse have occurred. Adjuvant radiation therapy may be used postoperatively once healing of the surgical site has been obtained.
Akin to multiple myeloma as a descendent of plasma cell malignancies, plasmacytoma is a solitary lesion that usually affects the vertebral body. Plasmacytomas generally affect younger patients than multiple myeloma and are associated with a better prognosis. Plasmacytomas eventually can evolve into multiple myeloma; thus, patients should be monitored for more than 20 years following the original diagnosis of plasmacytoma.
The diagnosis is made by biopsy of the lesion, and treatment includes radiation and bracing except in persons with pathologic or impending pathologic fractures. In these individuals, surgical resection and stabilization should be carried out with postoperative adjuvant radiation therapy once 6-8 weeks of postoperative healing has occurred. Patients have greater than 60% 5-year survival rates.
The tumors that most commonly metastasize to the spine are as follows:
Tatsui et al found that patients with prostate cancer had the highest rate of metastases to the spine.[22] They also found that lung cancer was the most common primary lesion in patients whose spinal metastases were detected before the diagnosis of primary lesions.
The time from diagnosis of the primary lesion to detection of the spinal metastasis wsa shown to be shortest in patients with lung cancer and longest in those with breast cancer. Patients with metastases from breast cancer or prostate cancer had the highest 1-year survival rate, whereas patients with metastases from lung or gastric cancer had the lowest 1-year survival rates.
Complications associated with spinal tumors can be divided into the following two groups:
Fan et al conducted a study to analyze complications following posterior vertebral column resection in patients with spinal tumors.[25] A total of 36 complications were reported, and the following associations were noted:
The majority of the complications were minor and did not affect patient recovery. The investigators concluded that active preventive measures are necessary to reduce the incidence of major complications.
Chang et al conducted a study to evaluate local control rate and to identify prognostic factors after stereotactic radiosurgery for treatment of primary malignant spinal tumors.[26] Median age of the 29 patients was 46 years (range, 11-68). Histologic diagnoses included chordoma (n = 11), chondrosarcoma (n = 5), osteosarcoma (n = 3), synovial sarcoma (n = 3), plasmacytoma (n = 2), Ewing sarcoma (n = 2), malignant peripheral nerve sheath tumor (n = 2), and malignant fibrous histiocytoma (n = 1). Mean follow-up was 50 months (range, 8-126).
Surgical resection was the initial treatment in 25 cases, percutaneous biopsy in four.[26] Stereotactic radiosurgery was used as primary treatment in 14 cases and as salvage treatment for progressive lesions in 15. Eleven patients had undergone previous conventional external-beam radiation therapy before stereotactic radiosurgery. Median tumor volume was 14 cm3 (range, 2.0–235). Delivered radiation doses were 12-50 Gy in 2-6 sessions. The mean radiation dose converted into a biologic effective dose (BED) was 60 Gy (range, 43–105).
Mean overall survival was 84 months for chordoma patients and 104 months for sarcoma patients.[26] The investigators found no factors that affected overall survival. The mean local progression-free survival was 56 months for chordoma patients and 73 months for sarcoma patients. The recurrent mode of presentation was predictive of local progression of spinal sarcomas. For patients with chordoma, no factors were found to correlate with local recurrence.
Kose et al conducted a study of the effect of early rehabilitation on neurofunctional outcome after surgery in children with spinal tumors.[27] The investigators reviewed medical charts and radiographic records of 70 pediatric patients (aged 1–17 years) who underwent surgery for the removal of spinal tumor. The patients received rehabilitation treatment beginning 4 days (range, 2–7 days) after surgery for 10 days (range, 7–23 days).
Results were assessed on the basis of scoring on the Modified McCormick Scale, the Functional Independence Measure for Children, the American Spinal Injury Association Impairment Scale, and the Karnofsky Performance Status Scale.[27] Sensory function, motor function, and activity of daily living were significantly improved for the patients who received early rehabilitation. Tumor setting, the level of localization, and the patients' clinical symptoms had no bearing on neurofunctional outcomes.
Copyright © www.orthopaedics.win Bone Health All Rights Reserved