Congenital deformities of the spine are spinal deformities identified at birth that are a byproduct of anomalous vertebral development in the embryo. Minor bony malformations of all types occur in up to 12% of the general population and are usually not apparent, often identified only on routine chest films or lumbar spine films (see the image below).[1] In contrast, congenital spinal malformations that result in progressive spinal deformity are relatively rare, occurring with a reported frequency of 0.5/1000 births.[2]
 Minor malformations of the spine seldom are apparent and often are identified only on routine chest films.
Minor malformations of the spine seldom are apparent and often are identified only on routine chest films.
Congenital anomalies of the spine have a range of clinical presentations. Some congenital abnormalities may be benign, causing no spinal deformity and remain undetected throughout a lifetime. Others may be associated with severe, progressive spinal deformity leading to cor pulmonale or even paraplegia (see the image below).
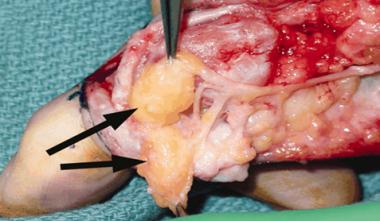
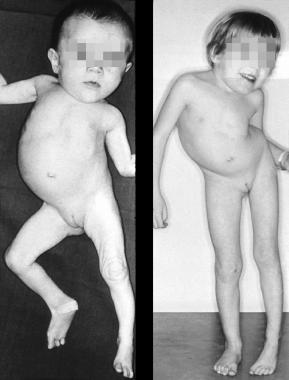 Clinical results of an untreated, unilateral unsegmented bar.
Clinical results of an untreated, unilateral unsegmented bar.
Congenital spinal deformity may be described broadly in terms of the direction of the particular deformity. Some deformities will result in sagittal plane abnormalities (kyphosis or lordosis), whereas others will primarily affect the coronal plane (scoliosis). The resultant spinal deformity is often a complex, three-dimensional structure with differences in both the coronal and sagittal plane, along with a rotational component along the axis of the spine.[3] (See the images below.)
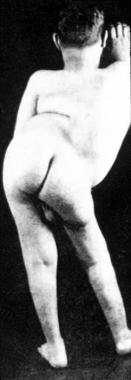 A patient with severe, congenital hyperlordosis.
A patient with severe, congenital hyperlordosis.
 Congenital kyphosis from a posterior, unbalanced hemivertebra.
Congenital kyphosis from a posterior, unbalanced hemivertebra.
For patient education resources, see the Osteoporosis Center, as well as Scoliosis.
A basic understanding of embryonic spine development is required to appreciate the patterns of malformation seen clinically and the associated malformations in other organ systems commonly identified.
Human development in utero (gestation) has been divided into the embryonic period and the fetal period. The embryonic period is considered to be the time from fertilization to the end of the week 8 of gestation. The remainder of gestation is called the fetal period. By the end of the embryonic period, all of the major organ systems have been established, and the basic body plan is complete.
In the early embryo, the mesoderm on either side of the neural tube (paraxial mesoderm) differentiates into paired blocks of cells called somites. The somites further differentiate into sclerotomes, groups of cells that ultimately give rise to the vertebral column and rib cage. The sclerotomal cells collect segmentally at the embryonic midline, surrounding the neural tube and the notochord, and form the precursors of the vertebral arch and vertebral body. (See the image below.)
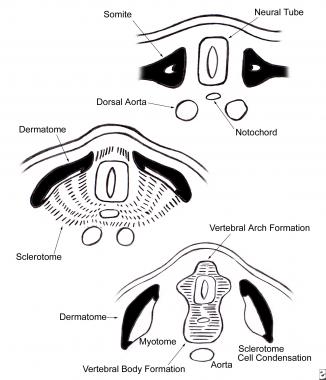 Ventromedial cells of the sclerotome migrate toward the midline of the embryo to surround the neural tube and the notochord. There they form the precursors of the vertebral arch and vertebral body.
Ventromedial cells of the sclerotome migrate toward the midline of the embryo to surround the neural tube and the notochord. There they form the precursors of the vertebral arch and vertebral body.
The sclerotomal cells also begin to separate into a cranial portion and a caudal portion. The cranial portion of each sclerotome recombines with the caudal portion of the directly superior sclerotome in a resegmentation process known as metameric shift. After the metameric shift, spinal nerves, which originally left the neural tube to go to the center of the sclerotome, are able to pass between the precartilaginous vertebral bodies to innervate the segmentation myotomes. (See the image below.)
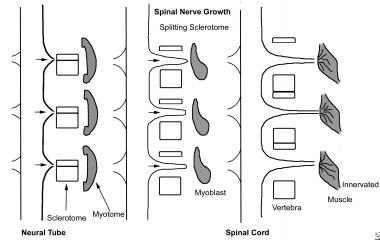 Vertebral morphogenesis; each vertebral sclerotome splits into a cranial and a caudal portion, which recombine with the superior and inferior sclerotome, permitting the segmental spinal nerves to grow out and to innervate the myotome derivatives.
Vertebral morphogenesis; each vertebral sclerotome splits into a cranial and a caudal portion, which recombine with the superior and inferior sclerotome, permitting the segmental spinal nerves to grow out and to innervate the myotome derivatives.
The atlas and axis form via a mechanism different from that governing the formation of the other vertebral bodies. Part of the first cervical sclerotome plus the cranial portion of the second cervical sclerotome contribute cells, forming the odontoid process and the arch of the atlas. In the cervical region, the eight cervical somites generate seven cervical vertebrae because the cranial portion of the first cervical sclerotome contributes to the formation of the occiput, and the caudal portion of the eighth cervical sclerotome contributes to T1.
In this way, the eight cervical spinal nerves become associated with seven cervical vertebrae (see the image below). The first cervical spinal nerve passes between the base of the skull and the first cervical vertebra. The eighth cervical nerve exits below the seventh cervical vertebra and above the first thoracic vertebra. The remainder of the nerve roots exit below their corresponding vertebral bodies.
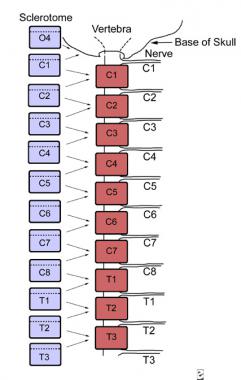 The cervical spine has 8 nerves as a result of the resegmentation of the sclerotome. The cranial portion of the first cervical sclerotome combines with the fourth occipital sclerotome to contribute to the base of the skull, whereas the eighth cranial somite contributes to C7 and T1.
The cervical spine has 8 nerves as a result of the resegmentation of the sclerotome. The cranial portion of the first cervical sclerotome combines with the fourth occipital sclerotome to contribute to the base of the skull, whereas the eighth cranial somite contributes to C7 and T1.
The intervertebral disks form in the area between the segmented vertebral bodies after the split of the sclerotomes. The nucleus pulposus originates from cells of the notochord, whereas the annulus fibrosis originates from the sclerotomal cell.
Alterations in the increasingly complex molecular and cellular tissue interactions of embryogenesis likely lead to structural defects involving the spine and spinal cord. Such defects may occur prenatally, postnatally, or both and can be grouped into the following three categories:
The majority of congenital spinal deformities may be understood in terms of malformations. However, it is useful to understand that the ultimate severity of the deformity is only partly attributable to the initial structural asymmetry imparted by the vertebral malformation and relates more to the persistent asymmetric growth of the malformed elements.
At the same time that the sclerotomes are forming the vertebral bodies, the heart, kidneys, trachea, and esophagus are differentiating. A noxious influence during this time can therefore affect the development of these organs as well. Hence, cardiac anomalies often are associated with congenital scoliosis of the thoracic spine, and renal anomalies are often associated with congenital malformation of the lumbar spine.
Intraspinal malformations (syrinx, diastematomyelia and tethered cord) are also found with congenital malformations of the bony spine. VACTERL syndrome (abnormalities of the v ertebrae, a nus, c ardiovascular tree, t rachea, e sophagus, r enal system, and l imb buds) is an extreme example of the associated malformations in synchronously developing organ systems.
The ultimate cause of a congenital spinal abnormality is an irrecoverable difference in spine development at the embryonic level. Much research has been performed to understand the mechanisms of embryonic segmentation, and a variety of genetic defects have been suggested as a cause for congenital vertebral malformations.
Single nucleotide polymorphisms in glucose metabolizing genes including GLUT1, HK1, and LEP have been linked to the occurrence of malformations observed in diabetic embryopathy.[1] Hox -mediated gene expression has been thought to be the target for spine abnormalities related to carbon monoxide.[4] In addition, an increased risk of congenital vertebral malformations is noted in monozygotic and dizygotic twins.[5]
Despite the isolation of genetic mechanisms, no convincing familial linkage exists to explain the majority of congenital scoliosis cases. Winter et al found that only 13 of 1250 patients had a positive family history of such deformity.[6] Furthermore, hereditary congenital scoliosis is relegated to sporadic case reports and is described mostly in the setting of an overlying syndrome, such as Jarcho-Levin (extensive defects of segmentation in association with spondylocostal, costovertebral, or spondylothoracic dysplasia).
The ultimate etiology is probably multifactorial, involving some combination of inherited genetic susceptibility and de novo alteration in molecular mechanisms, possibly from exposure to teratogens such as cigarette smoking, organophosphate pesticides or carbon monoxide.
(See also Kyphosis, Scheuermann Kyphosis, Idiopathic Scoliosis, Infantile Scoliosis, Neuromuscular Scoliosis, and Klippel-Feil Syndrome.)
NextCongenital scoliosis is a lateral curvature of the spine that is caused by congenital anomalies of vertebral development. The vertebral abnormalities are present at birth, but clinical deformity may not be evident until later in childhood, when progressive scoliosis is evident. The male-to-female ratio for congenital scoliosis is 1:1.4.[7, 8, 9]
This type of scoliosis must not be confused with infantile idiopathic scoliosis. Although the latter also can present as a deformity in early childhood, spinal radiographs show that there are no vertebral anomalies in the infantile form of idiopathic scoliosis.
Congenital spinal deformity is classified according to the types of anomalies. The following classification of MacEwen, advocated by the Scoliosis Research Society, has been well accepted:
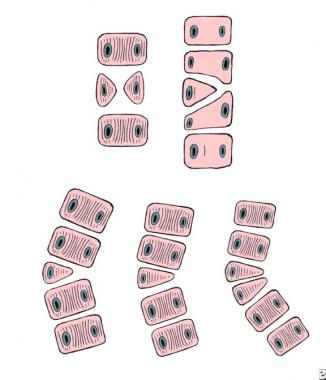 Failure of formation. Top left: anterior central defect. Top right: incarcerated hemivertebra. Bottom, from left to right: free hemivertebra, wedge vertebra, and multiple hemivertebrae.
Failure of formation. Top left: anterior central defect. Top right: incarcerated hemivertebra. Bottom, from left to right: free hemivertebra, wedge vertebra, and multiple hemivertebrae.
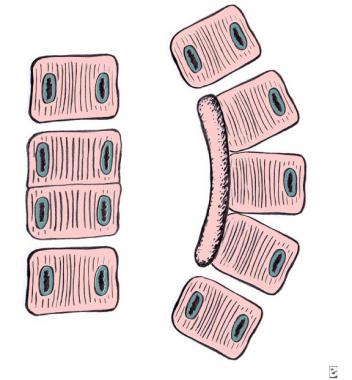 Failure of segmentation. Left: block vertebra. Right: unilateral unsegmented bar.
Failure of segmentation. Left: block vertebra. Right: unilateral unsegmented bar.
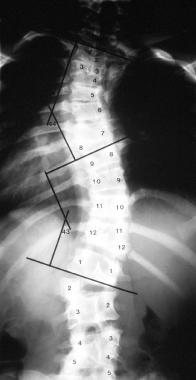 Mixed vertebral deformity involving the thoracolumbar spine.
Mixed vertebral deformity involving the thoracolumbar spine.
Defects of formation may be classified as follows:
The scoliosis that develops may occur with kyphosis or lordosis, depending on the precise location of the defects. Specific defects of segmentation may be classified as follows:
Variations of hemivertebrae are common, and prognosis depends on specific patterns. Hemivertebrae may be subclassified as follows:
Knowledge of the natural history of these congenital deformities is essential because the natural history dictates the prognosis and treatment. The natural history of congenital scoliosis has been described in several excellent studies. The large study by McMaster and Ohtsuka is the best in this regard.[10] They found that only 11% of cases were nonprogressive, whereas 14% were slightly progressive, and the remaining 75% were significantly progressive.
The prognosis for congenital scoliosis with regard to its rate of deterioration and final severity depends on many factors, including the following:
The type of anomaly that causes the most severe scoliosis is a unilateral unsegmented bar with contralateral hemivertebrae at the same level. Next in severity is a scoliosis caused by a unilateral unsegmented bar alone, followed by two unilateral fully segmented hemivertebrae, a single fully segmented hemivertebra, and a wedge vertebra. The least severe scoliosis is caused by a block vertebra.
Congenital scoliosis caused by unclassifiable anomalies can be difficult to predict and requires careful monitoring. A poor prognosis is associated with a unilateral unsegmented bar with or without contralateral hemivertebra, which should be treated immediately, without a period of observation (see Table 1 below).
Table 1. Vertebral Anomalies Leading to Congenital Scoliosis (Open Table in a new window)
Risk of Progression (Highest to Lowest) Curve Progression Unilateral unsegmented bar with contralateral hemivertebrae Rapid and relentless Unilateral unsegmented bar Rapid Fully segmented hemivertebra Steady Partially segmented hemivertebra Less rapid Incarcerated hemivertebra May slowly progress Unsegmented hemivertebra Little progressionAs regards the site of the anomaly, the rate of deterioration of the resulting scoliosis is most severe in the thoracic and thoracolumbar regions and usually is less severe in the cervicothoracic and lumbar regions. On the other hand, a mild cervicothoracic curve may produce an unsightly appearance because of the head and neck tilt and the elevation of the shoulder line. Lumbar curves do not cause much cosmetic deformity unless decompensation or pelvic obliquity occurs.
Congenital scoliosis, like idiopathic scoliosis, tends to progress most rapidly during the preadolescent growth spurt, after age 10 years. Scoliosis presenting as a clinical deformity in the first few years of life has a particularly bad prognosis because this indicates a marked growth imbalance that will continue throughout the period of growth, resulting in severe deformity.
In general, multiple balanced anomalies throughout the spine do not progress, and the cosmetic appearance is satisfactory. The more unbalanced the anomalies, the more likely the scoliosis is to progress.
Because of the high frequency of associated anomalies within and outside the spine, evaluation of a patient who has a congenital spinal deformity is very different from that of a patient who has a more common idiopathic or neuromuscular spinal deformity. Letts et al found that 82% of patients with congenital scoliosis had associated malformation in 4 different organ systems.[11] Beals et al found that 61% of patients with congenital scoliosis had abnormalities in seven different organ systems. Highest on the list were anomalies of the genitourinary tract.
Research on patients with congenital scoliosis by MacEwen et al revealed a 20% incidence of urinary tract anomalies detected on routine intravenous pyelography, whereas a study by Hensinger et al on cervical spine anomalies found a rate of 33%.[12, 13] Many of these anomalies, such as the presence of a single kidney, duplicate ureters, or crossed renal ectopia, while being of interest, were not potentially dangerous. However, in about 5% of the patients, obstructive uropathy, most commonly urethrovesicular obstruction, was present.
Renal ultrasonography and magnetic resonance imaging (MRI) may be used to diagnose renal anomalies accurately.[14]
A second area of great concern is cardiac anomalies. As many as 10-15% of patients with congenital scoliosis have been noted to have congenital heart defects. Murmurs should never be attributed to the scoliosis and must be evaluated thoroughly.
The frequency of spinal dysraphism is high in patients with congenital scoliosis. The prevalence of a dysraphic lesion was approximately 40% in three independent studies. McMaster reported that about 20% of these patients with congenital scoliosis had some form of dysraphism (eg, diastematomyelia, tethered spinal cord, fibrous dural band, syringomyelia, or intradural lipoma). Many other anomalies can occur in addition to the above problems, such as Sprengel deformity, Klippel-Feil deformity, Goldenhar syndrome (oculoauriculovertebral dysplasia), and anal atresia.
Clinical evaluation
Taking a full, detailed history and performing a full physical examination are mandatory because associated anomalies of many organs are common. Maternal perinatal history, family history, and developmental milestones must be explored fully. A comprehensive review of systems includes evaluation for the following:
In the physical examination, the physician must not only explore the spinal deformity but also focus particular attention on chest deformities and cutaneous lesions (especially dimples and hair patches overlying the spine). A detailed neurologic examination should be performed.
The genitalia should be examined for maturity, epispadias, hypospadias, and the presence of undescended testicles. The hand must be examined for clubhand, thenar hypoplasia, or other, more subtle, anomalies. The feet must be studied for clubfeet, cavus or varus deformities, vertical tali, clawing of the toes, or other signs of motor weakness.
The spine is best examined with the patient fully disrobed and erect. Skin dimples and hair patches should be documented. Shoulder and pelvic levelness are assessed, and asymmetry and prominence of the scapulae are noted. With the patient forward flexed, rotational deformity is recorded as centimeters of rib hump, and any lack of spinal motion is documented.
Radiographic evaluation
An accurate radiologic evaluation is essential. Standard posteroanterior (PA) and lateral views of the entire spine are used for the initial evaluation. Standing films are customary because they show the characteristics of the curve under gravity, with its compensation and torso-pelvic relationships. Supine, coned-down views of the anomalous region are especially helpful for visualizing defects and their patterns, while bending films allow evaluation of rigidity or flexibility of the curves and their adjacent motion segments.
Convex growth is important.[15] Therefore, the quality of the bone and disk spaces on the convexity must be clearly visualized and inspected. If the disk spaces are present and clearly defined and the convex pedicles clearly formed, convex growth is possible, and the prognosis is poor. If the convex disks are not clearly formed and the convex pedicles are poorly demarcated, less convex growth potential is present, and the prognosis is not as bad.
In the first 1-2 years of life, cartilage forms a significant part of the vertebrae; thus, at this stage, prognostication is not as accurate as it is later in childhood. Tomography (if available) is a useful technique for evaluating congenital spinal deformities, especially for documenting the presence and extent of unsegmented bars and incarceration of hemivertebrae.
Computed tomography (CT) and MRI are particularly useful in detailing bony canal anatomy and associated spinal cord dysraphism. These evaluations are mandatory prior to surgical intervention because spinal cord tethering and diastematomyelia must be identified and released prior to correction of the curve.[14, 16]
The primary goal of treatment of congenital scoliosis is to prevent the development of a severe deformity, rather than waiting until a severe deformity has developed and then attempting to perform a major and dangerous corrective procedure.
For patients with a marked spinal growth imbalance, no treatment is perfect.[15] The best result that can be achieved is spinal growth that is balanced on the convexity. In these circumstances, the optimum result is a short, relatively straight spine rather than the severely crooked spine that would have developed without treatment. The following three factors are key in achieving optimum results in patients with congenital scoliosis:
If the diagnosis is made early, while the curvature is still small, an opportunity exists for prophylactic surgery to balance the growth of the spine.
The prognosis for deterioration of congenital scoliosis can be anticipated on the basis of the amount of spinal growth remaining, the type and site of the vertebral anomaly, and the degree of growth imbalance that it produces. This requires careful study of good-quality spinal radiographs and knowledge of the natural history of the condition.
It is easier to prevent a severe spinal deformity than to correct one. A unilateral unsegmented bar, with or without contralateral hemivertebrae, is associated with such a bad prognosis that no period of observation is necessary: Immediate surgical treatment is required, no matter how young the patient is.
Other types of congenital scoliosis may be observed, but one of the most common errors is the failure to recognize slow and relentless progression until it is too late for prophylactic treatment. Therefore, all patients require radiologic assessment at 4- to 6-month intervals; once progression is established, immediate treatment is necessary to prevent further deterioration.
Observation
The most common nonoperative management in congenital scoliosis is mere observation, monitoring the curves for progression. Patients are observed in cases in which the natural history is not known. No role for mere observation exists in a patient with an unsegmented bar, because this condition uniformly progresses, but mere observation does have a role in the management of hemivertebrae and of mixed deformities.
Radiographs are obtained at regular visits (every 4-6 months). The same vertebrae are measured on each radiograph, using exactly the same vertebral landmarks, and the current film is compared with the film from the last visit and the original film. These comparisons are necessary because progression is slow. The two periods of rapid growth are during the first 4 years of life and during adolescence (the adolescent growth spurt). More frequent visits are necessary during these periods.
Orthotic treatment
Unlike idiopathic scoliosis, for which bracing can be effective, very few cases of congenital scoliosis lend themselves well to brace treatment, because the primary deformity in congenital scoliosis is in the vertebrae rather than in the soft tissues, and the curves tend to be rigid. In addition, in cases in which the natural history indicates a poor prognosis, orthotic treatment is contraindicated. Thus, contraindications to orthotic treatment are as follows:
Congenital scoliosis responds well to bracing only when the scoliosis features long curves with good flexibility—best determined by bending or by traction radiographs—or when the scoliosis is unbalanced secondary to an unbalanced hemivertebra at the T12 or L5 level.
The brace of choice is the Milwaukee brace for high thoracic curves (apex T6 or above), because it avoids the constriction of the thorax that may occur with an underarm brace, and the thoracolumbosacral orthosis (TLSO) for lower curves. Winter et al indicated that certain patients did well in the Milwaukee brace for many years and that a few could even be treated permanently with an orthosis and avoid surgery.[17, 6, 18, 19, 20, 21]
The best results were in patients with mixed anomalies that were flexible and in patients with a progressive secondary curve. Braces are unlikely to be effective if the scoliosis is more than 40° or if less than 50% flexibility is established using side bending or distraction radiographs.[22, 23]
A significant shoulder elevation is best treated with a shoulder ring that is attached to the Milwaukee brace, and head support pads can be added to create a neutral head position if the patient has a head tilt. Only the Milwaukee brace and its modifications can control high curves.
The physician must recognize the role of the orthosis and must carefully monitor the curves clinically and radiographically. If the patient's curve progresses despite the orthosis, fusion must be performed without further delay. Orthosis treatment should be continued only if the orthosis successfully controls the curve. After surgery, however, a brace may be required to help control spinal alignment and the development of compensatory curves that were not included in the fusion.
No other known form of nonoperative treatment has any effect on congenital scoliosis. Exercises, manipulation, and electrical stimulators have completely failed.
Surgery is the most effective treatment for severe or progressive congenital scoliosis. Many different forms of operative treatment exist for congenital scoliosis, and all have their place. No single operative procedure can be applied to all types of deformities. The method of surgery selected depends on the age of the patient, the site and type of vertebral anomaly, the size of the curvature, and the presence of other congenital anomalies. Successful surgical treatment depends on selecting the right procedure and applying it at the right time.[24, 11, 19, 25, 26, 27, 28, 29, 30]
Progressive curves should be treated surgically, especially if they do not respond to orthotic treatment. For example, if a 25° curve in a 3-year-old child progresses to 35° by age 6 years, the curve requires surgical treatment. The tendency is to avoid surgery at this age for fear of stunting the child's growth, but in reality, the child will grow taller if the curve is fused than if a progressive deformity occurs, because the anomalies are devoid of normal vertical growth potential.
There are four basic procedures for the surgical treatment of congenital scoliosis[31, 32] :
The fusion can be in situ or with correction by traction, bracing, or instrumentation, although instrumentation should be used only to maintain correction and not to produce correction.
In a retrospective cohort study by Vitale et al, pulmonary function and quality of life were measured in 21 children treated with early spinal fusion for progressive congenital scoliosis. The patients were divided into two groups: those receiving thoracic fusion and those receiving nonthoracic fusion. The patients who underwent thoracic fusion had shorter spines, worse pulmonary function, and more pain, results that might support consideration of alternatives such as growing rods, epiphysiodesis, or distraction thoracoplasty.[33]
Convex growth arrest
The prophylactic convex growth-arrest procedure was first described by MacLennan in 1922 and subsequently was described by Roaf and others.[34, 25] It was designed to arrest the excessive convex growth and allow the concave growth to occur and correct the deformity. Therefore, it is indicated in cases with a progressive scoliosis or marked scoliosis with single or adjacent convex hemivertebrae and a chance for concave growth with normal or near-normal concave growth plates.
This procedure is best applied to a patient younger than 5 years when there is growth potential on the concavity, as evidenced by healthy-appearing growth plates, and a short curve of less than 60°. Theoretically, this should allow the scoliosis to slowly correct by means of the continuing growth on the concavity.
Convex growth arrest is a relatively safe procedure. The only disadvantage is the slow and often uncertain correction because of the unpredictable growth potential on the concavity of the curve. However, even if the deformity does not correct at all, a convex growth arrest often is sufficient to stabilize the deformity. This procedure is contraindicated if any kyphosis is present in the area of the anomaly, because the anterior portion of the fusion may aggravate the kyphosis.
The procedure is performed in two stages, usually under the same anesthesia. The spine is first approached anteriorly on the convexity of the scoliosis. The lateral half of the disks and their adjacent endplates are removed at the site of the hemivertebra and at one intervertebral level above and below. This removes the anterior growth plates at the site of the anomaly, which is the main cause of the increasing scoliosis. In older or larger children, it may be possible to perform this procedure thoracoscopically, avoiding a thoracotomy for thoracic level curves.
The second stage of the procedure is performed through a separate, posterior exposure of the convexity of the curve at the site of the hemivertebra. The paraspinal muscles on the concavity of the curve should not be stripped. A posterior convex fusion is performed to one level above the hemivertebra and to one level below it.
The child is placed in a postoperative body cast, which is trimmed in such a way that the chest tube (inserted more anteriorly than normal) can be removed from under the cast. The child is kept nonambulatory for 3-4 months, at which time the cast is removed and the child placed in a well-fitting spinal orthosis that is worn full time for 12-18 months. This protection is necessary in all cases of fusion in the young child. Reports of this combined growth-arrest procedure show that the best results are achieved with surgery at a younger age and with lumbar anomalies.
Posterior fusion
The aim of a posterior fusion is not curve correction but, rather, curve stabilization with the prevention of further curve increase. Posterior spine fusion without instrumentation is the basic criterion standard for congenital scoliosis surgery, but since the 1990s, the combined anterior and posterior approach has gained more common use.
Posterior fusion is the usual surgical procedure for an older child with a moderately severe curve that still is relatively flexible. The fusion must cover the entire measured curve and extend to the central gravity line. Although it usually is not possible to obtain correction at the site of the anomalous vertebrae, moderate correction may be achieved at the relatively normal vertebral levels that lie above and below this area and are still within the scoliosis.
The child should be immobilized completely with an appropriate molded Risser cast or with a well-fitting Milwaukee brace until the fusion is absolutely solid. Although a soft fusion may occur in 4-6 months, a strongly trabeculated fusion will not occur for about 1 year, particularly if bank bone has been used, because it takes longer to incorporate into the body.
The use of posterior spinal instrumentation to correct a congenital scoliosis at the time of the spinal fusion has several advantages. Posterior spinal instrumentation achieves a moderately better correction and a lower incidence of pseudoarthrosis than a posterior spinal fusion in a Risser jacket alone. Spinal instrumentation is, however, associated with a greater risk of neurologic complications because of the effect of distraction on the spinal cord while the patient is anesthetized.
A preoperative MRI scan is necessary when instrumentation is planned, to exclude any spinal dysraphism. Although spinal cord monitoring using evoked potentials is essential, it is not completely reliable.
The wake-up test should be performed immediately following correction of the deformity. This test monitors only the current neurologic status, and neurologic abnormalities can develop at a later stage in the operative procedure and even in a delayed manner postoperatively. Because many types of instrumentation are available today, the surgeon should choose the system that best fits the child, the deformity, the safety factors, and the surgeon's experience.
Combined anterior and posterior fusion
Combined anterior and posterior fusion is increasingly performed for congenital scoliosis. It is used for thoracic, thoracolumbar, or lumbar curves with a poor prognosis (those with good convex growth potential).
The multiple diskectomies provide an anterior growth arrest, which reduces or eliminates any bending of the fusion (crankshaft effect). This is far less frequent in congenital scoliosis than in juvenile idiopathic scoliosis because of the very abnormal growth plates and more rigid curvature. In addition, owing to the combined approach, the pseudoarthrosis rate is lower. This usually is performed under the same anesthetic.
It is preferable to perform the anterior procedure first, by removing an appropriate rib to expose the area of the curvature and then by removing the disk and growth plates. This provides greater mobility in the curvature, allowing a better correction.
The removed rib is placed in a trough that has been created in the vertebral bodies with a rongeur, and the periosteum is resutured. This provides a greater assurance of solid fusion without pseudoarthrosis. A thoracoscopic procedure can also be used in older children. A chest tube is inserted, the chest is closed, the patient is turned prone, and the standard posterior spine fusion is performed with or without instrumentation, depending on the circumstances.
The only circumstance in which anterior fusions are contraindicated is in the very young child with a kyphotic deformity, in whom the anterior growth plates are maintained to continue growth in the face of the solidly fused spine posteriorly.
Hemivertebra excision
Hemivertebra excision was first performed in 1921, by Royle in Australia.[26] Despite the long period of experience with the procedure that ensued, it was many decades before more than a very few reports existed of significant follow-up. The only exception was the excellent paper by Leatherman and Dickson.[35]
Excision of a fully segmented hemivertebra is theoretically attractive as a prophylactic procedure because it removes the primary cause of the scoliosis, which is the enlarging wedge of the convexity at the apex of the curve. Remember that the entire curve must always be fused after the wedge osteotomy.[36]
Hemivertebra excision is indicated for patients younger than 5 years who have developed a structural secondary curve with a fixed decompensation that cannot adequately be aligned using other procedures. This problem usually is due to a hemivertebra at the fourth or fifth lumbar level, because no spine exists below the hemivertebra to allow compensation. Hemivertebra excision is safer in this area than in the thoracic region because the cauda equina is more tolerant of manipulation than is the spinal cord.
The hemivertebra is excised in two stages. The spine is approached anteriorly and posteriorly in a manner similar to that for a convex growth arrest. In the first stage, the hemivertebral body and the anterior body of the pedicle are removed, as are the adjacent disks and vertebral endplates.
In the second stage of the procedure, the posterior elements of the hemivertebra are removed, along with the transverse process and posterior part of the pedicle. This is a technically demanding procedure with a risk of direct injury to the spinal cord. It may also interfere with the segmental blood supply to the spinal cord. Some surgeons have advocated separating the two stages by 10 days to allow time for the vasculature to recover. Others have reported successful results when both procedures were performed under the same anesthetic.
Lazar and Hall have described the simultaneous anterior and posterior hemivertebra excision technique with posterior instrumentation, using infant-sized Danek hardware. Once the hemivertebra has been excised, the scoliosis can be corrected by closing the wedge osteotomy. Depending on the age of the child and the size of the vertebrae, instrumentation is added, usually a compression system on the convexity of the excised segment, using hooks or pedicle screws.[37]
When it is not possible to add instrumentation, correction is maintained by using a body cast with a leg extension, with the child usually being nonambulatory for 3-4 months. With the use of internal fixation, the child is placed in a brace with a leg extension for 4 months, with additional immobilization if the fusion is not solid. Excision of a lumbosacral hemivertebra is contraindicated if a second hemivertebra is present on the opposite side in the lumbar region; excision of either hemivertebra further unbalances the spine and increases the deformity.
Vertebrectomy
Vertebrectomy is the most radical of all procedures for congenital scoliosis. It consists of the removal of two or more vertebrae in their entirety, including the pedicles from both sides, as well as the laminae, and bodies. Vertebrectomy is performed to create mobility, but it is carried out at the price of instability. The procedure is neurologically risky and must be accompanied by appropriate spinal cord monitoring and wake-up tests. It should be reserved for the most severe deformities and performed only by highly skilled spinal surgeons.
Congenital kyphosis is less common than congenital scoliosis but can, if left untreated, cause paraplegia. It exists in the following two types:
Defects of segmentation occur most often in midthoracic or thoracolumbar regions and may involve two to eight levels. They tend to produce a round kyphosis rather than a sharp, angular gibbous deformity; therefore, paraplegia rarely is a problem. The main clinical symptom is low back pain caused by the necessary compensatory lumbar hyperlordosis. Commonly, the kyphosis caused by the defect of segmentation starts in the late juvenile years with the progressive ossification of the disk space anteriorly.
In the early stages, differentiation from Scheuermann disease can be difficult, but with time, the progressive anterior ossification becomes obvious. The rate of progression is less than that with a formation failure because the bar forms in the late juvenile years and the growth discrepancy is not as great.
Defects of formation are more common and may involve only a single level, but multiple defects are possible. The failure of the formation can be purely anterior, resulting in kyphosis, or it can be anterolateral with a posterior corner hemivertebra, resulting in kyphoscoliosis.[38] The more severe the anterior defects, the more progressive the deformity.
Kyphosis from failure of formation is universally progressive and, if left untreated, can lead to paraplegia. The paraplegia may occur early, but it is more common during the adolescent growth spurt, with rapid increase in the untreated kyphosis; it may occur after minor trauma. Paraplegia is more common with kyphosis in the upper thoracic area because this is the part of the spinal cord with the poorest collateral circulation (the so-called watershed area of the spinal cord blood supply). In North America, congenital kyphosis is the most common cause of spinal deformity.
No role exists for nonoperative treatment of congenital kyphosis. All forms of nonoperative treatment, including full-time bracing, are useless. The natural history indicates a universally poor prognosis; therefore, treatment must be surgical.
Defects of segmentation
The choice of treatment depends on the magnitude of the deformity and whether correction is desired. If discovered early, defects of segmentation can be treated with a posterior fusion, which should include the entire kyphosis, as well as one vertebra that is cephalad to the lesion and one that is caudad to it. Because the deformity is rigid, instrumentation is not used unless the congenital kyphosis is part of a longer kyphosis that requires treatment and fusion.
When the kyphosis presents later with a significant deformity that requires correction, the combined approach is best, with multiple osteotomies of the anterior bar of the bone followed by posterior fusion. For larger patients, compression instrumentation is desirable. If the patient is too small for instrumentation, hyperextension casting is needed to maintain the correction until the area of the arthrodesis has fused.
Defects of formation
The main goal of treatment is prevention of paraplegia. If the defect is detected early, when the patient is younger than 5 years and when less than 50° of the kyphosis is yet present, posterior fusion alone will produce a desired outcome.
Postoperatively, the child is placed in a hyperextension cast and is kept nonambulatory for 3-4 months. In patients younger than 18 months or in those in whom a pseudoarthrosis is detected, a repeat posterior procedure is performed to reinforce the fusion or to repair the pseudoarthrosis. To achieve a good result, it generally is recommended that routine exploration and graft augmentation be performed at 4-6 months so that a thick fusion can be obtained.
Following reinforcement, the child is immobilized in a brace or, preferably, a hyperextension cast for 4-6 months. In their review of 17 cases of congenital kyphosis, in which posterior fusion alone was used in children younger than 5 years, Winter and Moe, with an average 9-year follow-up, found improvement in 12 patients (71%).
When the deformity is greater than 50° or when the child is older than 5 years, a combined anterior and posterior arthrodesis is mandatory. The anterior fusion is performed first, with radical excision of the anterior longitudinal ligament, the disks, and the related ligaments. If possible, a distractor should be used to lengthen the anterior column, and a structural bone graft from the rib or fibula should be placed anteriorly to maintain the achieved height.
The anterior procedure is followed (either on the same day or a week after) by a posterior arthrodesis and compression instrumentation if bones are large enough to accept hooks and rods. Otherwise, the patient is managed with a hyperextension cast and bed rest for several months.
With the use of instrumentation and secure fixation, the child can be ambulatory without immobilization. If the fixation is not secured or is in question, a cast or brace is best. Traction has no role in the treatment of congenital kyphosis, because traction in these cases is associated with a high incidence of paraplegia.
Patients who have neurologic deficits at the time of presentation require special consideration. All such patients should undergo MRI of the spinal canal.
In patients who present with minor neurologic deficits associated with congenital kyphosis (eg, spasticity manifested by hyperactive reflexes and positive Babinski signs, without loss of motor, bowel, or bladder function), an anterior decompression of the spinal cord is not necessary. The anterior and posterior fusion should be carried out as described previously, and the neurologic deficit will disappear gradually once the spinal canal has been realigned and the area has been stabilized.
In cases with more marked neurologic loss, the anterior and posterior arthrodesis must be combined with an anterior decompression of the spinal cord. The patient is kept nonambulatory for 2-4 weeks to allow maximal cord recovery and to permit postoperative edema in and around the cord to subside.
Congenital lordosis is the least common of the three major patterns of congenital spinal deformity. It is caused by a failure of posterior segmentation in the presence of anterior active growth. Pure congenital lordosis is rare, and congenital lordosis with failure of posterior formation is extremely rare. Asymmetrical defects of segmentation, such as a posterolateral, unsegmented bar leading to lordoscoliosis, are more common. Some authors have not seen any patients in whom congenital lordosis was caused by a defect of formation.
Congenital lordosis deformity usually is progressive. With an increasing lordosis deformity in the thoracic spine, the spine-sternal distance (or anteroposterior diameter of the chest) is reduced and the rib mechanics of respiration are altered, with resultant respiratory restriction, respiratory failure, and even early death. When the deformity occurs in the lumbar spine, it results in hyperlordosis, with the spine approaching the anterior abdominal wall.
Treatment of congenital lordosis is purely surgical. Nonsurgical treatment has no role, because the condition is progressive. Because the deforming force in these cases is anterior growth, all cases require an anterior approach. However, because these patients have pulmonary restrictive disease, an anterior approach has greater risks.[39] If an element of pulmonary failure is already present, these risks increase.
If the patient is suffering from pulmonary artery hypertension, surgery probably is contraindicated because of the high mortality in these cases. Two types of surgery—one for anterior fusion and another for correction — are available for congenital lordosis.
Anterior fusion
Anterior fusion can be performed when the surgeon has the opportunity to see the patient early, before major deformity and loss of pulmonary function have developed. Anterior fusion involves disk excision, removal of the cartilage endplates, and packing of the disk spaces with bone chips. The anterior fusion should include the entire involved area and one or two vertebrae cephalad and caudad to the lesion. This approach eliminates the anterior growth potential and provides an anterior fusion opposite the unsegmented bar.
Corrective surgery
A so-called corrective operation is performed when the patient has a major deformity and, usually, loss of pulmonary function.[39] The goal is to improve spinal alignment and pulmonary function.
A combined anterior and posterior approach is indicated in these cases. Anteriorly, the disks are excised over the whole area of the deformity, and thin wedges of the adjacent vertebral endplates are excised, converting the disk excision into an osteotomy that is wider anteriorly. The disk spaces are not packed with bone chips because the wedges need to close anteriorly.
The posterior portion of the operation consists of multiple osteotomies of the laminar synostosis, performed at the same levels from which the disks were excised anteriorly. The best correction method employs the passage of sublaminar wires; when the bones are large enough, these wires are used to approximate the spine to a kyphotically contoured rod (Harrington, Luque, or one of the third-generation multiple hook-rod systems).
Whenever feasible, a combined procedure should be performed during the same anesthetic session in order to gain an immediate increase in lung volume and vital capacity.
Sacral agenesis is the term that is commonly applied to a group of disorders characterized by absence of the variable portion of the caudal portion of the spine. An uncommon congenital deformity, it occurs in approximately 1 in 25,000 live births.
Patients with sacral agenesis lack motor function below the normal portion of the spine, much as persons with myelomeningocele do.[40] In addition, sensory function is impaired below the level of the affected vertebrae. In more severe cases of sacral agenesis, part or all of the lumbar spine and even the lower thoracic spine may be absent. Some authors use the term lumbosacral agenesis in these severe cases.
Hohl first described agenesis of the lower spine in 1852, and Friedel redescribed it in 1910. Since then, a number of review articles and case reports have appeared in the literature. Williams and Nixon appear to have been the first to use the term sacral agenesis.[41] Because the absence of the coccyx is not known to cause any problems, discussion of this condition seems to be unnecessary.
Renshaw classified patients according to the amount of sacrum remaining and the characteristics of the articulation between the spine and the pelvis (see the image below).[42]
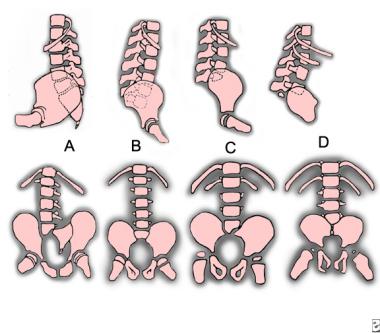 Sacral agenesis classification. A: type I. B: type II. C: type III. D: type IV.
Sacral agenesis classification. A: type I. B: type II. C: type III. D: type IV.
The classification types are as follows:
The exact etiology of sacral agenesis is unknown. In the human embryo, differentiation of the lumbar spine, sacrum, and coccyx occurs between the fourth and seventh postovulatory weeks. Duraiswami demonstrated that insulin injected into chick embryos can cause rumplessness, a condition similar to sacral agenesis.[43] Blumel et al first called attention to the increased frequency of diabetes in mothers of affected children, the incidence being about 16%.[44]
Although maternal diabetes is the risk factor that is most commonly associated with sacral agenesis, less common risk factors also have been described. Several reports have suggested that exposure to organic solvents in early pregnancy may increase the incidence of sacral agenesis.
The patient's physical appearance depends on the extent of the spinal involvement and on the degree of neurologic deficit. The characteristics of patients with sacral agenesis vary according to type, as follows.
Type I
In type I, the vertebropelvic articulation is usually stable. The unilateral absence of the sacrum results in an oblique lumbosacral joint and lumbar scoliosis. The scoliosis generally is not progressive and does not require surgical treatment. Hips and knees usually are normal. A calcaneovarus deformity of the foot may be present. Sensory loss corresponds to the distribution of the involved sacral roots.
Type II
In type II, the vertebropelvic junction is stable unless associated myelomeningocele is present.[40] Associated congenital anomalies of the spine, such as hemivertebrae, may cause progressive congenital scoliosis.
Motor paralysis is present and corresponds to within one level of the vertebral defect. Sensation usually is intact. Anesthesia may be present at S4, as well as distally. In patients with myelomeningocele, the level of paralysis may be higher than the level of vertebral deficit, and the sensory loss may be more extreme.[40]
Hip dislocation occurs in this type of sacral agenesis and may be unilateral or bilateral. Foot and knee deformities usually are not marked. Most patients with type II lumbosacral agenesis are ambulatory.
Type III
In type III, the lumbopelvic junction is relatively stable. Progressive kyphosis and scoliosis may develop in these patients, particularly when associated myelomeningocele is present. The level of the motor paralysis corresponds to within one segment level of the vertebral deficit. Sensation is intact at least down to the fourth sacral nerve root level. In cases of total absence of the sacrum, the buttocks are flattened, the cleft is shortened, and each buttock is dimpled lateral to the cleft. The normal convexity of the sacrococcygeal region is lost.
Hip dislocation, knee contracture, and foot deformity are common and require treatment. Patients with type III sacral agenesis are unable to stand or walk without appropriate support.
Type IV
In type IV, where the lumbar spine and sacrum are completely absent, patients are of short stature. The thorax and the pelvis are markedly disproportionate. The normal convexity of the sacrococcygeal region is lost, and the anus is horizontal. The pelvis is very unstable under the spine and tends to roll up under the thorax and drop forward. Almost all patients with type IV lumbosacral agenesis develop progressive kyphosis and scoliosis. They require surgical treatment for stabilization.
The hips have severe flexion and abduction contracture and may be dislocated. The knees show flexion contracture and large, popliteal webs. Fixed calcaneus deformities of the feet are present. Complete muscle paralysis and atrophy are present at and below the knees. Sensation usually is normal down to the knees. These patients have no bladder or bowel control. Patients with type IV sacral agenesis require spinal-pelvic stabilization or extensive orthotic support to ambulate.
High-level sacral agenesis presents the orthopedic surgeon with two unique problems: spinal-pelvic instability and severe knee flexion contractures, with popliteal webbing of the knee. Reconstruction of the lower limb in type IV sacral agenesis has not been successful, because of the absence of muscle fibers and major motor nerves.
Some authors have reported the results of treating patients with bilateral subtrochanteric amputation. These patients, fitted with a pelvic-thoracic bucket and a Canadian hip disarticulation prosthesis, were able to become partially ambulatory through the employment of a swing-to or swing-through gait, as used by persons with paraplegia.
As long as spinal-pelvic instability remains uncorrected, the patient is unable to sit without support or to ambulate without the aid of a pelvic-thoracic bucket. Many authors have performed spinal-pelvic fusion. Their justification for using the procedure was the need to protect the viscera from nonphysiologic compression and angulation, and to establish a stable vertebral-pelvic complex about which lower extremity contractures could be stretched or surgically released.
Currently, spinal-pelvic fusion does not seem justified for asymptomatic spinal-pelvic instability. If the spinal-pelvic fusion is to be performed, Winter's technique is recommended.[20] Autogenous tibiae (if knee disarticulation was performed) or allografts can be used for spinal-pelvic fusion.
Often, the knee flexion and foot deformities are corrected at an early age via surgical release. A combination of soft-tissue releases, supracondylar femoral extension osteotomies, and serial casting probably will yield satisfactory correction of some knee contractures. If amputation is chosen, disarticulation at the knee is the recommended level of amputation. The neuromuscular deficit is less in type I and type II sacral agenesis. These patients have spinal-pelvic stability, and they can sit and walk. The foot and knee deformities should be corrected.
Congenital anomalies of the cervical spine, though rare, are worthy of attention because neurologic compromise from instability or stenosis may be prevented with early recognition and careful management of persons who are at risk. Congenital anomalies range in severity from those that are benign and asymptomatic to anomalies that can potentially cause fatal instability.
Many anomalies are not discovered until a complication occurs. Anomalies of the occipitocervical junction often are not detected until late childhood or adolescence, and some remain hidden well into adult life. Other anomalies of the cervical spine, although recognized in early life, may not become clinically significant until adulthood. Although they are rare, it is important that such anomalies be recognized in order to prevent catastrophic paralysis as a result of sports participation or manipulation during anesthesia.
Basilar impression (or invagination) is a deformity of the bones of the base of the skull at the margin of the foramen magnum. The floor of the skull appears to be indented by the upper cervical spine; therefore, the tip of the odontoid is more cephalad. This increases the risk of neurologic damage from injury, circulatory embarrassment, or impairment of the cerebrospinal fluid (CSF) flow. The following types of basilar impression exist:
With basilar impression, the upper cervical spine encroaches on the brainstem and spinal cord as the base of the skull is displaced toward the cranial vault. Motor and sensory disturbances are noted in 85% of individuals who are symptomatic. However, most affected patients remain asymptomatic until the second or third decade of life, when they may present with headache, neck ache, and neurologic compromise; the prevalence of symptoms is 15%.
Basilar impression is difficult to assess radiographically, and many measurement schemes have been proposed. Those most commonly referred to are Chamberlain's, McGregor's, and McRae's lines on the lateral radiograph (see the image below), and the Fischgold-Metzger line on the anteroposterior projection.[45, 46, 47, 48]
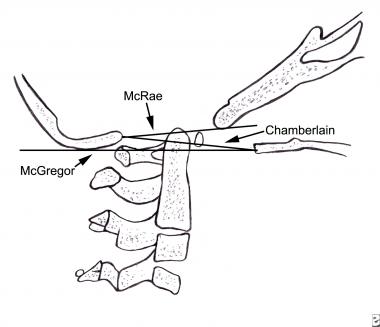 Lateral craniotomy. The drawing indicates the 3 lines used to determine basilar impressions on lateral radiographs.
Lateral craniotomy. The drawing indicates the 3 lines used to determine basilar impressions on lateral radiographs.
Chamberlain's line is drawn from the opisthion, on the posterior lip of the foramen magnum, to the dorsal margin of the hard palate. McGregor's line is drawn from the upper surface of the posterior edge of the hard palate to the most caudad point of the occipital curve of the skull. McRae's line defines the opening of the foramen magnum. McGregor's line is the best method for screening because the bony landmarks can be defined clearly in persons of all ages on a routine lateral radiograph.
The position of the tip of the odontoid is measured in relation to this baseline; a distance of 4.5 mm above McGregor's line is considered to be on the extreme edge of reference ranges. More detailed techniques (CT and MRI) generally are reserved for patients in whom a routine examination or clinical findings suggest an occipitocervical anomaly.
Treatment depends on the cause of the symptoms and often requires the combined effort of an orthopedic surgeon and a neurosurgeon. Anterior impingement from a hypermobile odontoid may require fusion in extension if the odontoid can be reduced. If the odontoid cannot be reduced, an anterior excision and stabilization in extension can be considered. Posterior impingement may require a suboccipital craniectomy and decompression of the posterior ring of C1 and possibly C2 with the release of tight dural bands. This is followed by fusion of the occiput to C2 or C3.
This malformation is characterized by a partial or complete congenital union between the atlas and the base of the occiput. The condition is also known as occipitalization of the atlas and assimilation of the atlas into the occipital bone.
Occipitocervical synostosis, basilar impression, and odontoid anomalies are the most common developmental malformations of the occipitocervical junction. Incidence ranges from 1.4-2.5 per 1000 children, and the 2 sexes are affected in equal numbers. The age range of patients presenting with this anomaly is reported to be 8-52 years. Some patients develop symptoms following mild trauma, while others remain entirely asymptomatic throughout life. Weakness and ataxia in the lower limbs and occasionally in the upper limbs are the primary symptoms.
The following are the most common signs and symptoms of occipitocervical synostosis, in decreasing order of frequency:
Clinical findings also include a low hairline, torticollis, a high scapula, and a short, broad neck, as well as restricted neck movements, such as those seen in Klippel-Feil syndrome. This disorder is commonly associated with C2-C3 fusion.
Other associated anomalies that are occasionally seen include dwarfism, funnel chest, pes cavus, syndactylies, jaw anomalies, cleft palate, congenital ear deformities, hypospadias, and, in some cases, genitourinary tract defects.
Standard radiographs of this area can be difficult to interpret. Tomography, CT, and MRI may be necessary to clarify the pathologic condition. Bony anomalies that may be visible include a backward tilt of the odontoid process, an articular facet between the anterior rim of the occiput and the odontoid process, asymmetrical atlantoaxial joints as seen on the anteroposterior view, and fusion of the body and lamina of the second cervical vertebra to the third cervical vertebra.
Initially, nonoperative measures in the form of traction and support of the neck by an orthosis are employed. If neural impairment is present, consideration may be given to decompression by laminectomy, craniectomy, and the release of posterior dural bands. The risk of morbidity and mortality is high.
Anomalies of the odontoid process, or dens, may range from complete absence (aplasia) of this feature to partial absence (hypoplasia) of the process to separation of the dens from the axis (a condition known as os odontoideum; see the image below). These anomalies can lead to atlantoaxial instability and may cause neurologic deficit and even death.
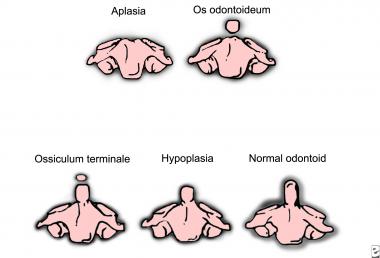 Gradations of the odontoid process's appearance.
Gradations of the odontoid process's appearance.
The frequency of these anomalies is unknown, and like many anomalies that may be asymptomatic, they probably are more common than is recognized. Aplasia is extremely rare, and hypoplasia and os odontoideum are reported infrequently and can be considered rare. Odontoid anomalies in association with ligamentous laxity, producing atlantoaxial instability, are more common in patients with Down syndrome, Morquio syndrome, Klippel-Feil syndrome, and some skeletal dysplasias.
Clinical presentation occurs, on average, in persons aged 19 years. Patients may be asymptomatic or may present with local neck symptoms, transitory episodes of paresis following trauma, or frank myelopathy secondary to cord compression. An important factor differentiating os odontoideum from other anomalies of the occipitovertebral junction is that patients with this condition seldom have symptoms referable to cranial nerves, because the area of the spinal cord impingement is below the foramen magnum.
In 40% of patients, the clinical manifestations are limited to neck pain and torticollis without neurologic involvement, and the prognosis is excellent. A few patients may have symptoms and signs of cerebral and brainstem ischemia, seizures, and mental retardation. Sudden death resulting from minor trauma has been reported.
Recommended radiographic views are open mouth, anteroposterior, lateral, flexion extension, or CT reconstructions. Flexion-extension CT scans are of value because plain films do not always show the anomaly or the extent of motion. At birth, the normal odontoid can be seen in the lateral view with the epiphyseal plate. A mistaken impression of hypoplasia may be given by a lateral extension radiograph.
Partial or complete absence of the dens can be congenital or acquired. This extremely rare anomaly may be recognized from birth onward and is best seen in the open-mouth view. The most common form of hypoplasia presents with a short, stubby peg of odontoid projecting just above the lateral facet articulations.
Tomography is necessary to confirm whether an os odontoideum is present in addition to the hypoplasia. CT is useful in patients with multiple anomalies; the usual radiographic views are not always reliable in confirming the presence or absence of an odontoid process.
In os odontoideum, there is a jointlike articulation (which appears radiologically as a wide, radiolucent gap) between the odontoid and the body of the axis. This gap may be confused with a normal finding in patients younger than 5 years. In children, therefore, the diagnosis is confirmed by demonstrating motion between the odontoid and the body of the axis.
Measurements can be made by using a line projected superiorly from the posterior border of the body of the axis to a line projected inferiorly from the posterior border of the anterior arch of the atlas. Measurements of more than 3 mm should be considered pathologic. MRI is helpful for evaluating the space available for the spinal cord.
Surgical stabilization is indicated if neurologic involvement is present, along with more than 10 mm of instability on supervised flexion-extension films; if instability is progressive; or if neck symptoms persist. The suggested method of stabilization is posterior cervical fusion of C1-C2, with wire fixation and an iliac bone graft. This is not without risk, because slight flexion often is required to pass the wire beneath the posterior ring of the atlas.
Operative reduction should be avoided; preoperative correction with traction or positioning is preferred. Prophylactic stabilization is controversial. However, good outcomes and low morbidity have been achieved with surgery.
Klippel-Feil syndrome is uncommon. As currently used, the term refers to all patients with congenital fusion of the cervical vertebrae, whether it involves two segments, congenital block vertebrae, or the entire cervical spine. Congenital cervical fusion is the result of failure of normal segmentation of the cervical somites during weeks 3-8 of life. With the exception of a few patients in whom this condition is inherited, the etiology is undetermined. The frequency is about 1 in 42,000 births, and 65% of patients are female (see the image below).
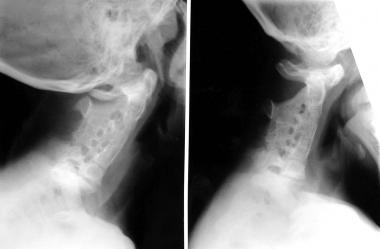 Klippel-Feil syndrome with congenital fusion of the entire cervical spine.
Klippel-Feil syndrome with congenital fusion of the entire cervical spine.
The classic clinical triad of low posterior hairline, short neck, and limitation of neck motion is seen in 40-50% of patients. The decrease in motion most commonly is in lateral bending and rotation. Except in the most massive fusion, flexion and extension are relatively well preserved. A low posterior hairline seems to reflect the shortening of the neck rather than being a separate entity.
The age at presentation is extremely variable, extending through the entire life span. Massive fusions often are noted in infancy or early childhood because of the resulting cosmetic deformity. Neurologic problems in infancy usually are related to craniovertebral junction abnormalities, although cervical vertebral fusions also may be present in this age group.
C1-C2 fusions often present in childhood, usually with pain. Lower cervical fusions, if they are not massive, often do not present until the third decade or even later in life, when degenerative changes or instability of adjacent segments develop.
Patients with Klippel-Feil syndrome present with a wide range of chief complaints, including cosmesis of the neck or cosmesis resulting from associated anomalies (such as Sprengel deformity), neck pain alone, radicular pain with or without weakness, slowly progressive or acute paraparesis or quadriparesis, and complications caused by associated anomalies. (See the list of associated anomalies below.)
Diagnosing congenital cervical fusion in infants and young children can be difficult. Flexion and extension can demonstrate a lack of motion between fused segments, and flexion and extension laminograms may be necessary to confirm the diagnosis. CT also can be used to define the bony architecture.
Aside from vertebral fusion, flattening (wasp waist) and widening of the involved vertebral bodies, as well as absent disk spaces, are the most common findings. The sagittal and transverse diameters of the spinal canal usually are normal. Narrowing of the spinal canal, if it occurs, usually is seen in adults and results from degenerative changes or hypermobility.
All of these defects may extend into the upper thoracic spine, particularly in cases of severe involvement. These disturbances may be detected first on routine chest film as an initial clue. With a high thoracic congenital scoliosis, the radiographic evaluation routinely should include lateral views of the cervical spine.
Common anomalies associated with Klippel-Feil syndrome, in decreasing order of frequency, include the following:
Anomalies less commonly associated with Klippel-Feil syndrome include the following:
The following patterns of deformity are at the greatest risk for instability and require early recognition:
Conservative treatment (eg, activity modification, bracing, and traction) may reduce symptoms. The indications for and timing of stabilization are not clearly defined. However, spinal fusion must be considered if a neurologic lesion is present, if significant pain persists despite conservative measures, or if instability is documented. Similarly, if neurologic injury is present and stenosis is documented, decompression with or without fusion is indicated.
Treatment of the cosmetic aspects of this deformity has met with limited success. Soft-tissue procedures, Z-plasty, and muscle resection may achieve cosmetic improvement in selected patients. All patients with congenital cervical fusion should avoid contact sports, as well as occupations and recreational activities that put them at risk for head trauma. The potential for catastrophic outcomes with this lesion must be kept in mind. Long-term follow-up should be undertaken, and patients and families should be counseled carefully.
Several syndromes are associated with congenital spinal deformity. Some of these are described here.
Down syndrome is characterized by hypotonia, a flat facies, slanted palpebral fissures, and small ears. Incomplete fusion of vertebral arches of the lower spine occurs in 37% of persons with Down syndrome, atlantoaxial instability in 12%, abnormal odontoid process in 6%, and a hypoplastic posterior C1 arch in 26%.
Any child with Down syndrome who develops changes in bowel or bladder function or in neck posturing or who loses ambulatory skills should be evaluated carefully with plain radiographs of the cervical spine. Most patients develop symptoms when they are younger than 10 years, when the ligamentous laxity is most severe.
Lejeune first described deletion 5p syndrome in 1963.[49] The common features are catlike cries in infancy, microcephaly, and a downward slant of the palpebral fissures. Hemivertebra and scoliosis are frequent occurrences.
Kabuki syndrome was described in Japan by Niikawa et al and by Kuroki et al.[50, 51] Skeletal abnormalities include a short, in-curved fifth finger secondary to short fourth and fifth metacarpals, as well as rib anomalies, brachydactyly, sagittal cleft of vertebral body, hip dislocation, and scoliosis. All cases have occurred sporadically in otherwise healthy families.
The first case of Noonan syndrome was described by Kobilinsky in 1883.[52] Noonan and Ehmke further delineated the clinical phenotype.[53] This syndrome is characterized by webbing of the neck, pectus excavatum, cryptorchidism, and pulmonary stenosis. Skeletal abnormalities include cubitus valgus, spina bifida, and hemivertebrae.
Described by Aarskog in 1970, Aarskog syndrome (also known as Aarskog-Scott syndrome) has become increasingly recognized.[54] The common features are hypertelorism, brachydactyly, and shawl scrotum. Skeletal abnormalities include broad thumbs and great toes, scoliosis, cubitus valgus, splayed toes with bulbous tips, and metatarsus adductus.
Wildervanck initially described cervico-oculo-acoustic syndrome in 1952. This disorder is characterized by Klippel-Feil anomaly (fusion of two or more cervical vertebrae and sometimes of thoracic vertebrae), abducens paralysis with retracted globs, and sensorineural deafness, torticollis, and Sprengel deformity.
Duncan described the MURCS association in 1979. It consists of a nonrandom association of müllerian duct aplasia, renal aplasia, and cervicothoracic somite dysplasia. The incidence of cervicothoracic vertebral defects, especially from C5-T1, is 80%. Other abnormalities may include Sprengel deformity, upper limb defects, and moderately frequent rib anomalies.
VACTERL syndrome consists of vertebral defects, anal atresia, cardiac anomalies, tracheoesophageal fistula with esophageal atresia, radial and renal dysplasia, and limb bud anomalies. Vertebral anomalies occur in 70% of cases. Other, less frequent defects are defects of the lower limb and spinal dysraphia with a tethered cord.
Jarcho and Levin described this disorder in 1938. It is inherited as an autosomal recessive condition, though autosomal dominant inheritance has been reported. Abnormalities include short-trunk dwarfism of prenatal onset, a short thorax with a crablike rib cage (in association with multiple vertebral and rib defects), posterior fusion and absence of ribs, lordosis, and kyphoscoliosis. Although most affected individuals die in early infancy as a result of recurrent pulmonary infection, a small number have lived beyond age 1 year.
Proteus syndrome is most commonly characterized by hemihypertrophy, subcutaneous tumors, and macrodactyly. Skeletal abnormalities include bony prominences over the skull, angulation defects of the knees, scoliosis, kyphosis, hip dislocation, dysplastic vertebrae, and clinodactyly.
Other examples of syndromes that are associated with congenital spinal deformity are arteriohepatic dysplasia, Gorlin syndrome, fibrodysplasia ossificans progressiva syndrome, Morquio syndrome, spondylo-carpo-tarsal synostosis syndrome, multiple synostosis syndrome, and Coffin-Lowry syndrome.
Copyright © www.orthopaedics.win Bone Health All Rights Reserved