A hamartoma (from Greek hamartia “fault, defect” and -oma, denoting a tumor or neoplasm) is a benign (noncancerous) tumorlike malformation made up of an abnormal mixture of cells and tissues found in areas of the body where growth occurs. It is considered a developmental error and can occur at a number of sites. A nonneoplastic mass can also arise in this way; therefore, misdiagnosis is possible, as is subsequent overtreatment with its added morbidity and mortality. Developmental remnants may be considered hamartomatous if they form discrete tumorlike masses.
The literature describes several examples of hamartomas, including the following:
A hamartoma resembles a neoplasm, but in most cases, it does not show any tendency to evolve into one. However, cases of neoplastic evolution have occurred with these lesions. Examples are chondrosarcomas arising in osteochondromas and neurofibrosarcomas arising in patients with von Recklinghausen disease.
In addition, neoplasms can be associated with hamartomas without directly arising from them. Examples are fibromas of the ovary and malignant ovarian tumors arising in patients with Peutz-Jeghers syndrome. Hamartomas should be distinguished from choristomas; the former are composed of tissues that are normally present at a given site, whereas the latter are malformations of tissues that are not normally found at that site.
Hamartomas may not cause any problems and are usually identified incidentally. Uncomplicated hamartomas have no tendency to grow, except as determined by the normal growth controls of the body. However, this does not mean that hamartomas are harmless. Morbidity can arise by means of a variety of mechanisms, including the following:
For additional, more detailed descriptions of most of the hamartomas described in this article, see the following:
For ease of description, the hamartomatous bone tumors are classified into the following four groups (see Table 1 below):
Table 1. Developmental/Hamartomatous Tumors (Open Table in a new window)
Lesion Class Tumors Bone-formingBone islands, also known as enostoses, are single areas of mature compact bone in cancellous bone. The lesions are developmental in origin, and they are usually incidental findings. A focal failure of bone resorption during skeletal maturation and remodeling is thought to result in the formation of these bone tumors.[1]
Osteopoikilosis consists of multiple bone islands. This disease is also known as osteopathia condensans disseminata or spotted bone disease. It is rare and is typically asymptomatic.
Melorheostosis is considered to be a mesodermal disorder of bone and soft tissue. It is a rare condition characterized by cortical thickening of unknown etiology. An irregular, sclerotic thickening that affects mainly the long bones is noted. The hallmark of this condition is its candle-wax appearance on radiographs.[2]
Osteopathia striata, also known as Voorhoeve disease, is a benign, asymptomatic disorder that shows symmetrical, opaque striations on radiography. When associated with osteopoikilosis and melorheostosis, osteopathia striata is called mixed sclerosing bone dystrophy.
Osteochondroma, also referred to as exostosis, is defined as a bony outgrowth capped by cartilage on the surface of the bone. It is the most common benign bone tumor. Osteochondroma is caused by herniation of the epiphyseal cartilage through a developmental defect in the epiphyseal plate.[3]
Multiple osteochondromas, also known as hereditary multiple cartilaginous exostoses, are usually associated with short stature and other bony deformities. The exostoses are rare. However, compared with solitary osteochondroma, exostoses are associated with a 1.3-6.0% increase in the risk of malignant transformation.[4]
Epiphyseal osteochondroma, also known as Trevor disease, is similar to a solitary osteochondroma. Patients present with unilateral enlargement of the epiphysis in an irregular way.
Enchondromatosis, also known as Ollier disease, is a rare developmental abnormality that usually manifests in childhood. This condition is characterized by a hamartomatous proliferation of cartilage in the metaphyses of several bones. It most commonly affects the hands and feet, causing distorted lengthwise growth or pathologic fractures. Chondrosarcoma develops in as many 50% of patients.[5]
Nonossifying fibroma, also known as a fibrous cortical defect or benign fibrous histiocytoma, is characterized by a solitary benign lesion occurring in the metaphysis of a long bone in a child, commonly the femur or the tibia. When the tumor grows into the medullary cavity and achieves the size of several centimeters, it is then referred to as a nonossifying fibroma.[6]
Fibrous dysplasia, also known as fibro-osseous dysplasia or fibro-osseous lesion, usually is solitary, slow-growing, and formed mainly of fibrous and bony tissue, though on occasion it may be composed of cartilage. If a coexisting soft tissue tumor is present, the condition is referred to as Mazabraud syndrome.
Hemangioma of bone, an irregular accumulation of blood vessels in bone, is usually solitary and asymptomatic. The skull and vertebral bodies are commonly involved, though any bone can be affected.
Skeletal hemangiomatosis and lymphangiomatosis—also known as cystic angiomatoses and lymphangiectases of bone, respectively—are incidental findings that can be associated with pathologic fractures, soft tissue masses, or effusions. They are often associated with cutaneous or visceral hemangiomas. Cystic angiomatosis may be considered a vascular hamartoma.
Bone islands usually occur in intramedullary spongy bone of the pelvis, femur, humerus, and ribs; they are uncommon in the thoracic or lumbar vertebrae of the spine. Bone islands are small, white-yellow, circular masses. Compact bone is seen in the haversian systems. Bony spicules arise from these masses and create a spokelike appearance in the trabecular bone. The usual size is 1-2 mm, but lesions as large as 10 cm have been documented. Lesions larger than 2 cm are termed giant bone islands.
Osteopoikilosis occurs most commonly in the small bones of the hands and feet, as well as at the ends of the long bones of the hands[7] ; it is rarely found in the skull, facial bones, ribs, or spine. The histologic appearance of osteopoikilosis is similar to that of bone islands.
The lesions of melorheostosis are commonly progressive and spread in a linear fashion. If they cross an open epiphyseal plate or a joint space, they can mimic osteochondromatosis. The progressive nature of these lesions is marked by increasing pain and deformity.
The pathophysiology of osteopathia striata is currently unknown.
Osteochondroma and multiple osteochondromas are large masses that may be sessile or (occasionally) pedunculated. Their size ranges from 1 to 10 cm, and their surface is smooth or lobulated and often covered by a thin fibrous layer. The bony protuberance has a thin cortex and a medullary cavity that is continuous with the affected bone. The osteochondroma is derived from abnormal cartilaginous epiphyseal growth plate tissue. It remains in subperiosteal and either disappears by means of remodeling or proliferates as an early osteochondroma.
The growth of osteochondromas terminates at puberty with the fusion of the epiphyseal plate. Osteochondromas that continue to grow after puberty should raise the possibility of malignant degeneration. Multiple osteochondromas have a relatively high (1-25%) incidence of malignant transformation.[4]
There are many theories regarding the pathophysiology of epiphyseal osteochondromas. The most common are abnormal regulation of cartilage proliferation,[8] a congenital error of epiphyseal development,[9] and a fetal developmental abnormality in the apical cap of the limb.[10]
Enchondromatosis is believed to arise from remnants of the epiphyseal cartilage. Growing slowly, these lesions erode the adjacent cortex and cause the contours of the bone to expand. Rarely do they rupture into the soft tissue. Foci of calcification or ossification may be present and punctuate; this is the classic osteolytic appearance seen on radiographs. Multiple large and eccentric cartilaginous tumors can appear in any part of the bone.
Nonossifying fibromas are common, benign, and well-delineated solitary lesions. They exist in the metaphysis of long bones. The tumors are thought to arise from the continued growth of fibrous cortical defects that extrude into the medullary cavity. The tibia and the femur are the bones most commonly involved. The lesions commonly regress and can result in new bone growth in areas of involution.[11]
Fibrous dysplasia usually manifests as solitary, slow-growing hamartomatous lesions. The overgrowth of fibrous bone leaves areas of bony weakness, which results in deformity through repetitive minor fractures. Repeated fracture of the femur results in the characteristic shepherd’s-crook deformity at the upper end of the bone.[12]
Although hemangiomas of bone are the most frequently occurring tumors in the vertebral body, they are usually incidental findings. Vertebral hemangiomas are located mostly at the thoracic level. Medullary hemangiomas are rare. When they occur, they are found in the metaphyseal region. Periosteal hemangiomas are also rare and involve the midshaft of long tubular bones. Finally, cortical hemangiomas of the long bones are rare, and most are in the tibial shaft.
The lesions are well-demarcated, dark red cavities with hamartomatous proliferation of the vascular tissue in the endothelial lining of the cavity. These lytic bone lesions are usually limited to the vertebral bodies, but they can cross the joint spaces. Patients can develop symptoms as a consequence of nerve compression or collapse of a vertebral body.
Skeletal hemangiomatosis is rare, and patients usually present in the first two decades of life; by comparison, lymphangiomatosis is most commonly identified in early childhood.[13] The lytic defects of skeletal hemangiomatosis can occur anywhere in the bone and give a honeycomb pattern. They can increase in size or regress, with eventual obliteration of the cystic cavity due to sclerosis.
Lymphangiomatosis involves the formation of cystic spaces, which are lined with a single layer of epithelial cells and filled with proteinaceous fluid. A combination of hemangiomas and lymphangiomas may also occur. The distinction is often made at the time of surgery. Skeletal hemangiomatosis and lymphangiomatosis are commonly self-limiting. Cases associated with involvement of additional soft tissue or viscera worsen the patient’s prognosis.
Bone islands are developmental in origin. No associated risk factors are reported.
Osteopoikilosis has an autosomal dominant pattern of inheritance. Associated conditions include Buschke-Ollendorff syndrome and other forms of sclerosing bone dysplasia (eg, melorheostosis and osteopathia striata).
The etiology of melorheostosis is unknown.
Osteopathia striata has an autosomal dominant pattern of inheritance when associated with sclerosis of the skull base.[14]
The etiology of osteochondroma is unknown. Evidence suggests an abnormality in the long arm of chromosome 8. After irradiation in childhood, the risk of the development of one or more of these tumors increases 10-12%.[15]
Multiple osteochondromas have an autosomal dominant pattern of inheritance. Three gene mutations have been identified in association with tumor suppressors EXT1 and EXT2. This disease is associated with other genetic syndromes, including Langer-Giedion syndrome and DEFECT 11 syndrome (also referred to as proximal 11p deletion syndrome or Potocki–Shaffer syndrome).
Epiphyseal osteochondroma is a rare developmental abnormality, and its etiology is unknown.
Enchondromatosis is a sporadic condition with no reported familial association. It is usually diagnosed before the age of 10 years. It is associated with Maffucci syndrome (enchondromas and hemangiomas) and metachondromatosis (enchondromas and hemangiomas); the former is nonhereditary, whereas the latter has autosomal dominant transmission. Enchondromatosis is associated with tumor-suppressor genes in the malignant transformation to chondrosarcomas. Notable alterations are inactivation of 9p21 and 13q14 and overexpression of p53.[16]
Nonossifying fibroma is a nonaggressive developmental abnormality with no known genetic predisposition. It often involves the metaphysis of long, growing bones, and it usually regresses spontaneously. Nonossifying fibroma can be an incidental finding on radiographs, or it may be secondary to a pathologic fracture. The observation that most lesions seem to occur near points of tendinous attachment suggests that the disease may be associated with stress or trauma.
With fibrous dysplasia a gene mutation is involved that results in the overgrowth of fibrous bone. The bone is weak, and this weakness predisposes the patient to pathologic fractures. No environmental or dietary factors have been associated with fibrous dysplasia, and the cause of the gene mutation is unknown. The condition begins before birth and is most commonly seen in childhood. The femur, tibia, skull, and ribs are the bones most commonly affected.
Fibrous dysplasia is usually monostotic and asymptomatic. When it is polyostotic, more than 60% of patients present before the age of 10 years. About 3% of cases are associated with McCune-Albright syndrome, and 50% are associated with abnormal cutaneous pigmentation. Other diseases that may be associated with fibrous dysplasia are desmoplastic fibroma, eosinophilic granuloma, giant cell reparative granuloma, and secondary aneurysmal bone cysts.
With hemangiomas of bone, asymptomatic lesions are usually discovered incidentally. No familial tendency is reported.
Skeletal hemangiomatosis or lymphangiomatosis is usually an incidental finding on radiographs. No familial tendency is reported.
The figures given below are for hamartoma frequencies in the United States. In most cases, international frequencies are similar to those in the United States, unless stated otherwise.
Bone islands are found in approximately 1% of the population and occur in adults more often than children. There is no sexual predilection, and no risk factors that predispose to the development of bone islands are known. This profile differs from that of osteoid osteomas, which are most commonly seen in male individuals and which make up 10-12% of all benign bone tumors. Likewise, osteosarcomas most commonly affect male persons; however, they are most frequent between the ages of 10 and 20 years.[17]
Seven hundred to 900 cases of osteosarcoma are diagnosed in the United States each year. About 400 of these cases are in persons younger than 20 years.[18] Osteosarcoma is the most common primary malignancy of bone in children.[19]
Osteopoikilosis demonstrates no sexual predilection, and it can appear at any age. The lesions are associated with a predisposition to form keloids, sclerodermalike lesions, and other subcutaneous nodules in 25% of patients.[20]
Melorheostosis is most common in the young and has no sexual predilection.
Osteopathia striata is a rare disease, one that is often associated with other bony hamartomatous tumors. Its incidence and prevalence are currently unknown.
Osteochondroma is most commonly seen in the second and third decades of life. Because most lesions are asymptomatic, the true prevalence is unknown. No sexual predilection has been established, though some authors report a 2:1 male-to-female preponderance.[21]
Multiple osteochondromas are commonly observed in childhood, with 40-50% of the lesions found in the distal femur and proximal tibia. This disease has an autosomal dominant mode of inheritance. It is not sex specific, but the proportion is increased among white persons.[22]
The incidence of epiphyseal osteochondroma is higher among males than among females, with a 3:1 ratio. When this disease occurs, it affects the medial aspect more often than the lateral aspect. The total estimated prevalence is 1 case per million population.[23]
Enchondromas account for 12-14% of benign bone neoplasms and 3-10% of neoplasms in general.[5] Malignant transformation occurs in 30% of patients,[24, 25] and 25% present with a sarcoma at the age of 40 years. The tumor most often associated with enchondromatosis is a chondrosarcoma (grade 1, grade 2, or dedifferentiated). Evidence also suggests an association with osteosarcomas and chordomas. When enchondromatosis is associated with systemic syndromes, the frequency of sarcoma reaches 30-50%.[26]
The exact incidence of nonossifying fibroma is unknown, but reports have suggested that 30-40% of all children may have one or more of these lesions at any one time.[27] The exact incidence is unknown, but an increased incidence has been reported in children, adolescents, and young adults aged 2-18; the male-to-female ratio is 2:1. The most commonly affected sites are in the lower extremity (femur, 40%; tibia, 40%; and fibula, 10%); the upper extremity is rarely involved.[28]
The exact incidence of fibrous dysplasia is currently unknown. This lesion is thought to account for 7% of all benign bone tumors. It occurs equally in male and female individuals. Approximately 75% of all cases are diagnosed before the age of 30 years.[29]
Different histologic types of hemangioma of bone exist. Vertebral hemangiomas account for 10-12% of all bone hemangiomas found at autopsy. About 75% of hemangiomas involve the vertebra and skull, 10% involve the long bones, and 5% involve the ribs. The male-to-female ratio is 2:1.[30]
Hemangiomas of bone account for 1% of all spinal neoplasms and represent 2-3% of all spinal tumors.[31] They are the most common benign tumor of the spinal column. The lesions occur most commonly in the lower thoracic vertebrae, less frequently in the lumbar spine, and infrequently in the cervical spine and sacrum.
Skeletal hemangiomatosis and lymphangiomatosis are usually discovered in childhood. The prevalence and incidence is currently unknown, because the diagnosis is often serendipitous. Solitary lesions are rare, and many cases of lymphangiomatosis of the bone are associated with visceral and soft tissue involvement. About 60% of all cases of skeletal hemangiomatosis involve other viscera, commonly the spleen. However, evidence in the literature also describes involvement of the liver, lungs, and soft tissue.
Bone islands are usually incidental findings on radiographs and do not normally cause symptoms. Patients with osteoid osteomas usually present with a dull, aching pain as the main symptom. Associated findings are swelling or a mass. Osteoblastomas usually clinically appear with pain lasting from 2 weeks to 1 year. The pain is described as boring, with localized tenderness. Osteosarcomas manifest with pain, swelling, and a palpable mass.
The differential diagnosis of bone islands can be made only with imaging. Differential diagnoses include calcified osteoid osteomas, osteoblastomas, osteoblastic metastases, and sclerotic osteosarcomas. Bone islands are also similar to osteopoikilotic lesions.
The presentation of osteopoikilosis is similar to that of bone islands; both conditions are asymptomatic. Differential diagnoses include bone islands and osteoblastic metastases from breast carcinoma.
Most patients with melorheostosis are asymptomatic. The differential diagnosis includes parosteal sarcoma if the disease is localized and includes myositis ossifications and calcified hematoma if the disease is associated with soft tissue calcification.
Osteopathiastriata is most commonly benign and asymptomatic. This condition is usually associated with osteopoikilosis and melorheostosis; when it is, it is referred to as a mixed sclerosing bone dystrophy. Differential diagnoses include osteopoikilosis and the autosomal dominant form of osteopetrosis.
Most patients with osteochondroma or multiple osteochondromas are asymptomatic, though they may also present with a palpable mass, pain, or a pathologic fracture. Evidence suggests an association with spinal cord compression, pseudoaneurysm formation, vein thrombosis, or bursitis. The differential diagnosis includes metachondromatosis, turret exostoses, and bizarre parostealosteochondromatous proliferation, among other rare syndromes.
Epiphyseal osteochondroma usually involves a single limb and occurs in young children. The disease most commonly occurs in the lower femur, upper tibia, or talus. Patients can present with unequal limb lengths, and valgus or varus deformities of the limbs may develop. The possibility of an epiphyseal osteochondroma should be kept in mind in pediatric cases when a bony lesion occurs near to a joint in the lower limb.[32] Differential diagnoses include chondroblastoma, osteochondroma, and enchondroma.
Patients with enchondromatosis usually present before the age of 10 years. The hands and feet may be involved. Severe defects of the femur can result in bowing and shortening of the bone, and patients can develop deformity of affected bones. The major differential diagnosis is chondrosarcoma.
Most nonossifying fibromas are asymptomatic. Large lesions can be symptomatic, however, and patients can present with a pathologic fracture. Jaffe-Campanacci syndrome is mainly characterized by nonossifying fibromas, extraskeletal congenital anomalies (eg, café-au-lait spots, mental retardation, hypogonadism, or cryptorchidism), and ocular and cardiovascular malformations.[33] The differential diagnosis includes fibrous dysplasia, osteoid osteoma, and chondromyxoid fibroma.
Monostotic forms of fibrous dysplasia are commonly asymptomatic. Patients may report slight pain, tenderness, swelling, or deformity. The disease process may become apparent because of a pathologic fracture.[29] Polyostotic forms are symptomatic before the age of 10 years. The incidence of pathologic fractures is increased in these cases. An abnormal cutaneous pigmentation is an associated finding in 50% of patients with polyostotic disease.
Approximately 3% of all polyostotic fibrous dysplasias are associated with McCune-Albright syndrome. Patients develop fibrous dysplasia, cutaneous pigmentation, associated endocrine disorders, acromegaly, hyperparathyroidism, Cushing syndrome. Girls often experience precocious puberty.
Differential diagnoses for fibrous dysplasia include enchondroma, enchondromatosis, eosinophilic granuloma, intraosseous osteosarcoma, giant cell tumor, bone hemangioma, nonossifying fibroma, primary hyperparathyroidism, Paget disease, and neurofibromatosis type 1.
Patients with hemangiomas of bone are asymptomatic; when they are symptomatic, the main clinical finding is progressive pain. Symptoms are associated with the site of occurrence. Examples are nerve compression, vertebral body collapse, and hematoma in association with vertebral hemangiomas. The differential diagnosis includes bone cysts, bone metastases, bone lymphomas, multiple myelomas, osteosarcomas, and Paget disease.
Hemangiomas of thoracic vertebra produce more symptoms that those within the lumbar vertebra. This is believed to be due to the less accommodative ratio of the spinal cord to the canal in the thoracic segment. This is particularly exaggerated in normal kyphosis of the thoracic spine.[34]
Reports in the literature state that pregnancy induces neurological symptoms from previously silent hemangiomas. It is believed to be caused by the gravid uterus causing venous obstruction, consequently leading to an increase in blood flow through the venous plexus. This results in expansion and growth of previously silent hemangiomas.[35, 36]
Patients with skeletal hemangiomatosis or lymphangiomatosis can be asymptomatic; the lesions can cause also pain, swelling, or a pathologic fracture.[37] The differential diagnosis includes familiar osteolysis, macro-osteolysis and essential osteolysis.[38]
Laboratory studies are not required unless patients are symptomatic. If needed, laboratory tests may include the following preoperative investigations:
Further investigations are unnecessary.
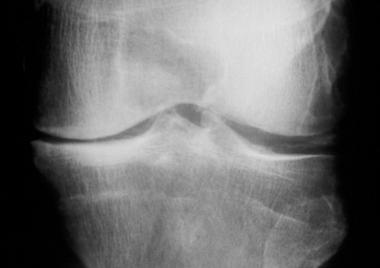
With bone islands, the results of basic investigations are inconclusive. As noted, these lesions are commonly incidental findings. On radiography, bone islands appear as round or ovoid areas with flecks of increased opacity in cancellous bone (see the image below).
 Bone island. On plain radiographs, bone islands typically appear as round-to-ovoid, sclerotic intramedullary foci. The long axis of the bone island is aligned parallel to the long axis of the bone.
Bone island. On plain radiographs, bone islands typically appear as round-to-ovoid, sclerotic intramedullary foci. The long axis of the bone island is aligned parallel to the long axis of the bone.
With osteopoikilosis, lesions are symmetrical and unequally distributed. They have a predilection to involve the epiphysis and metaphysis of long tubular bones. Radiographs show numerous discrete and homogeneous areas of increased radiopacity that are 3-5 mm in diameter. Findings are similar to those observed with bone islands (see the image below).
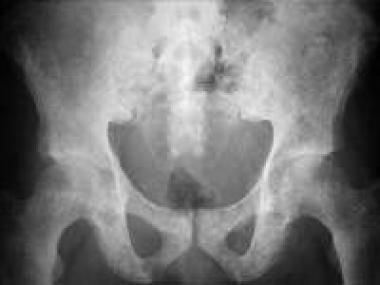 Osteopoikilosis. Plain radiograph of the pelvis shows numerous bone islands characteristic of osteopoikilosis.
Osteopoikilosis. Plain radiograph of the pelvis shows numerous bone islands characteristic of osteopoikilosis.
With melorheostosis, radiographs show well-defined lesions with undulating contours and a linear pattern of distribution (see the image below).
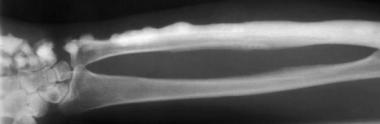 Melorheostosis. Image vividly depicts the characteristic flowing-wax or flowing-hyperostosis appearance.
Melorheostosis. Image vividly depicts the characteristic flowing-wax or flowing-hyperostosis appearance.
With osteopathia striata, lesions are often seen as bilateral linear bands of increased radiopacity on radiography, with interspersed areas of rarefaction (see the image below).
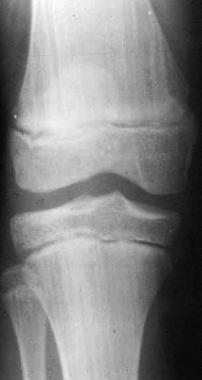 Osteopathia striata. This condition is characterized by longitudinal striations, which often appear at the metaphysis.
Osteopathia striata. This condition is characterized by longitudinal striations, which often appear at the metaphysis.
With osteochondroma and multiple osteochondromas, radiographs depict pedunculated or sessile masses with well-defined margins. When the tumors are pedunculated, the stalk points away from the adjacent joint surface. The cortex and the medullary cavity of the osteochondroma and the adjacent bone are continuous (see the image below).
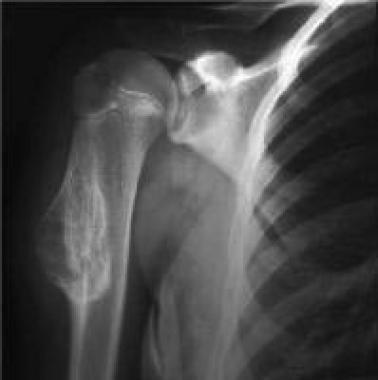 Solitary osteochondroma. Anteroposterior radiograph shows sessile osteochondroma of the humerus. See Image 6.
Solitary osteochondroma. Anteroposterior radiograph shows sessile osteochondroma of the humerus. See Image 6.
With epiphyseal osteochondroma and enchondromatosis, radiographs demonstrate radiolucent, often expansile lesions with a varied cartilaginous matrix and smooth or lobulated margins (see the images below).
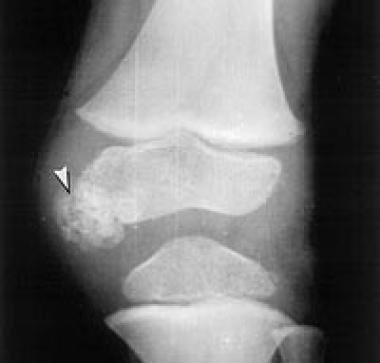 Epiphyseal osteochondroma. Image shows the anteroposterior knee of a 6-year-old boy. Characteristic lobular ossific masses are protruding from the medial distal femoral epiphysis (arrowhead).
Epiphyseal osteochondroma. Image shows the anteroposterior knee of a 6-year-old boy. Characteristic lobular ossific masses are protruding from the medial distal femoral epiphysis (arrowhead).
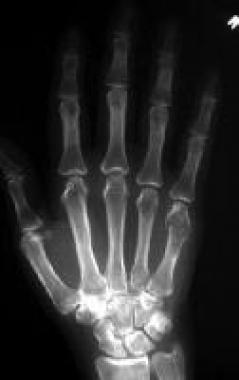 Solitary enchondroma. Frontal radiograph of the right hand demonstrates a lytic expansile lesion in the fifth metacarpal bone, with thinning of the cortex that has a somewhat scalloped appearance. A pathologic fracture is noted, but no calcifications are seen in the lesion. See Image 9.
Solitary enchondroma. Frontal radiograph of the right hand demonstrates a lytic expansile lesion in the fifth metacarpal bone, with thinning of the cortex that has a somewhat scalloped appearance. A pathologic fracture is noted, but no calcifications are seen in the lesion. See Image 9.
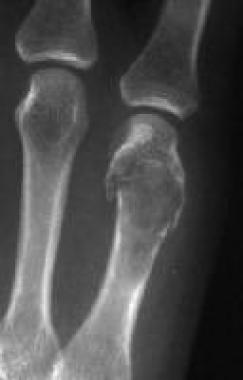 Solitary enchondroma. Detail of the lytic expansile lesion in the fifth metacarpal bone in the right hand depicted in Image 8. This image shows thinning of the cortex, with a somewhat scalloped appearance. A pathologic fracture is noted, but no calcifications are seen in the lesion.
Solitary enchondroma. Detail of the lytic expansile lesion in the fifth metacarpal bone in the right hand depicted in Image 8. This image shows thinning of the cortex, with a somewhat scalloped appearance. A pathologic fracture is noted, but no calcifications are seen in the lesion.
With nonossifying fibroma, radiographs show oval radiolucent lesions with ridged trabeculations in the bony wall and sclerotic borders. These lesions can also become involuted, resulting in the classic ground-glass or sclerotic appearance; this finding obviates further imaging.
With fibrous dysplasia, radiographic findings are the same in monostotic or polyostotic disease. Solitary lesions are usually small. Radiographs often depict centrally located, round, and expansile lesions. The lesions are either radiolucent or have a ground-glass appearance, which is usually due to new bone formation. The margins can be sharply defined or sclerotic (see the images below).
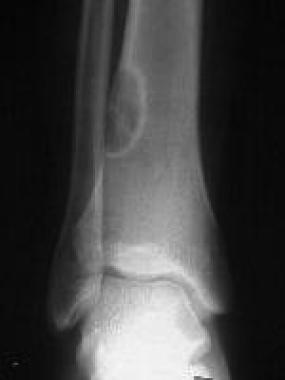 Fibrous cortical defect and nonossifying fibroma. Anteroposterior radiograph of the distal tibia shows a lobulated, well-circumscribed nonossifying fibroma that is eccentrically located in the distal tibia metadiaphysis. The peripheral sclerotic border with a central lucency is typical of this lesion.
Fibrous cortical defect and nonossifying fibroma. Anteroposterior radiograph of the distal tibia shows a lobulated, well-circumscribed nonossifying fibroma that is eccentrically located in the distal tibia metadiaphysis. The peripheral sclerotic border with a central lucency is typical of this lesion.
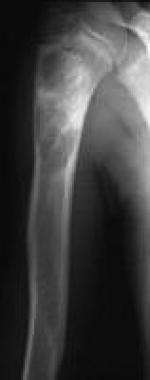 Fibrous dysplasia. Image shows homogeneous loss of the normal trabecular pattern in the shaft of the humerus, with a ground-glass appearance.
Fibrous dysplasia. Image shows homogeneous loss of the normal trabecular pattern in the shaft of the humerus, with a ground-glass appearance.
With hemangioma of bone or skeletal hemangiomatosis or lymphangiomatosis, radiographs show vertical striations reflecting the thickened trabeculae of the vertebral bodies. These cause the characteristic corduroy or honeycomb pattern appearance (see the image below).
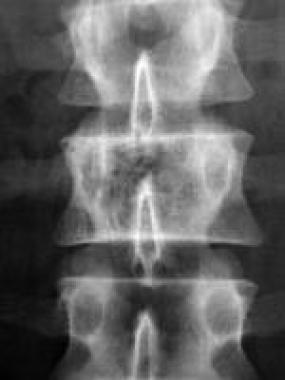 Bone hemangioma. Radiograph depicts the typical corduroy or accordion appearance of coarse, thickened vertical trabeculae in a hemangioma affecting the right side of the vertebral body at L2.
Bone hemangioma. Radiograph depicts the typical corduroy or accordion appearance of coarse, thickened vertical trabeculae in a hemangioma affecting the right side of the vertebral body at L2.
With bone islands, computed tomography (CT) shows no alteration in the contour of the bone. On magnetic resonance imaging (MRI), the compactness of the bone results in low signal intensity in cancellous bone on T1- and T2-weighted images. Bone scans usually show no activity; however, increased uptake has been documented in the literature.
With osteopoikilosis, bone scans usually depict no increase in uptake, though cases of uptake are reported in the literature.
With melorheostosis, CT scans highlight thickening of the cortex. On MRI, the lesions have the same features as those of cortical bone. Bone scintigraphy shows increased bone activity.[39]
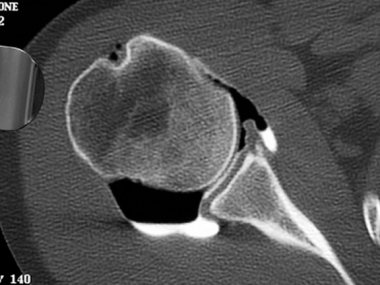
With osteochondroma and multiple osteochondromas, CT is useful for delineating osteochondromas in complex bones, such as those of the pelvis, shoulder, or spine (see the image below). MRI and ultrasonography are useful for evaluating the thickness of the cartilage cap. Bone scans are useful for differentiating benign from malignant osteochondromas; a normal scan practically eliminates the likelihood of malignancy. Angiography is relevant if the patient has vascular complications.
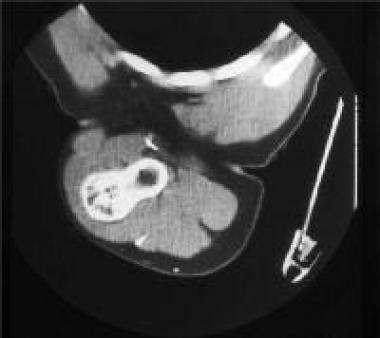 Solitary osteochondroma. CT scan of the sessile osteochondroma of the humerus shown in Image 5.
Solitary osteochondroma. CT scan of the sessile osteochondroma of the humerus shown in Image 5.
With epiphyseal osteochondroma and enchondromatosis, CT scans often highlight the extent of the lesion.[40]
With nonossifying fibroma, CT and MRI are used only to delineate cortical involvement in doubtful cases. Radionuclide imaging shows low-grade activity secondary to the new bone formation surrounding the lesion.
Initially, nonossifying fibromas have a high or intermediate T2 signal with a low signal rim corresponding to a sclerotic border. As the lesions matures and ossifies, the signal changes to become low signal.[41, 42]
With fibrous dysplasia, CT and MRI are often unnecessary, but the images can highlight cystic degeneration, aneurysmal formation, and cortical bony destruction. Bone scintigraphy and arteriography also yield positive results in most cases.
With hemangioma of bone and skeletal hemangiomatosis and lymphangiomatosis, CT scans show a coarsened appearance. On MRI, both T1- and T2-weighted images show increased signal intensity. MRI is the criterion standard for investigating vertebral hemangioma. Active lesions generate a low T1 and a high T2 signal, and quiescent lesions generate both high T1 and T2 signals. It is imperative that vertebral hemangioma be distinguished from giant cell tumors of the spine, metastatic lesions, and aneurysmal bone cysts.[43, 44, 45]
Angiograms depict a prominent tumoral blush. Radionuclide bone scans do not always yield positive results; however, they may be of value in locating multiple lesions. Lymphography often demonstrates abnormal and dilated vessels filling the bony lesions.
They are the most common benign tumor of the spinal column, usually within the vertebral body.[34] The lesions occur most commonly in the lower thoracic vertebrae, less frequently in the lumbar spine, and infrequently in the cervical spine and sacrum.[46, 47]
Characteristic histologic findings of hamartomas are as follows:
Because most hamartomas are found incidentally, their treatment depends on the patient’s signs and symptoms at the time of presentation. Pain is a common finding and can be treated conservatively with analgesics. If symptoms are not controlled, further investigation is usually required and surgical management is considered.
Most patients may not require any intervention. However, for those who do require intervention, procedures may include biopsy, curettage, and fracture fixation. If bony involvement is extensive, amputation of digits or limbs may result.
Regardless of the lesion or mass present, clinicians must adhere to certain basic principles. Whenever possible, benign tumors should be treated conservatively. Surgical intervention may be indicated in the following situations[48] ):
For patients undergoing surgery, the preoperative workup and management are similar for most hamartomatous lesions (see Laboratory Studies, Plain Radiography, and Other Imaging Studies). Specific intraoperative details are beyond the scope of this article; however, indications for surgical treatment of various hamartomas are briefly summarized below.
Specific postoperative details are also beyond the scope of this article. Follow-up care can range from no further intervention and simple reassurance to 6-month or annual visits, depending on the type of hamartoma and on the risk of malignant change.
Bone island
A bone island is important as a differential diagnosis. If this lesion enlarges, biopsy may be necessary. If the lesion is becoming symptomatic or if the patient has a history of malignancy, biopsy is indicated.
When radiographs demonstrate findings characteristic of a bone island, no follow-up is required, because the lesions are benign and are not associated with morbidity or mortality. Follow-up or biopsy may be indicated if a lesion is especially large, if it grows rapidly, or if it is found in a patient with symptoms or a history of malignancy that could produce osteoblastic metastases.[49]
Osteopoikilosis
The lesions of osteopoikilosis are benign and asymptomatic and do not alter morbidity or mortality. Some lesions may increase or decrease in size and number, or they may even disappear completely.[49] Despite osteopoikilosis being a benign disease, patients with osteopoikilosis should be monitored. Numerous case reports have demonstrated pathologies associated with osteopoikilosis, including spinal canal stenosis, hip fracture, and malignant degeneration.[50, 51, 52, 53]
Melorheostosis
The course of melorheostosis is insidious, with a slow, chronic progression of symptoms and periodic exacerbations. As melorheostosis becomes progressive, pain management is often the toughest challenge. Successful pain control can be achieved with the use of nonsteroidal anti-inflammatory drugs (NSAIDs) and bisphosphonates. However, such medications do not alter the course of bone lesion formation.[54, 55, 56, 57] Most cases are benign. In severe cases, lengthening of tendons or correction of deformities may be warranted, and amputation may be indicated.[58]
Osteopathiastriata
In and of itself, osteopathiastriata is benign and asymptomatic, and patients have a normal life expectancy. Although the combination of osteopathiastriata, macrocephaly, and cranial sclerosis is rare, it can cause disfigurement and disability secondary to pressure on the cranial nerves. Sequelae may include facial nerve palsy, deafness, and reduction of the visual field resulting from narrowing of the optic foramina.
With osteopoikilosis, melorheostosis, and osteopathiastriata, the relatively constant size and symmetrical distribution of the lesions make them obvious. If the disease is associated with subcutaneous nodules, they may be excised for cosmetic reasons. Surgery is indicated if patients become symptomatic. Evidence in the literature describes osteosarcoma formation.[59] When any of these conditions is associated with Buschke-Ollendorff syndrome, surgery may be required for cosmetic reasons.
Osteochondroma and multiple osteochondromas
Symptomatic patients with osteochondroma or multiple osteochondromas require surgical treatment. The specific presentation determines the urgency with which surgery must be performed. Patients with vascular ischemia, spinal cord compression, or rapidly progressive malignant degeneration require surgery on an urgent basis.
The prognosis and long-term survival of patients with osteochondroma or multiple osteochondromas vary greatly. In general, the prognosis is favorable because most osteochondromas cease to grow when skeletal maturity is achieved. When lesions continue to grow in adulthood, surgery is usually curative. One study showed that surgery resolved preoperative symptoms associated with osteochondromas in 93.4% of cases.[60] Failure to remove the entire cartilaginous cap or its overlying periosteum is the basis for most recurrences.
If a high-grade cancer grows, the outcome is somewhat uncertain. In rare cases, osteochondromas have spontaneously resolved during childhood or puberty. Continuous follow-up care is essential for children with multiple osteochondromas. Patients and their families should be informed about the hereditary nature of the condition.
Symptomatic patients with epiphyseal osteochondroma also require surgical treatment. Early excision is recommended to avoid intra-articular development, which would limit joint motion.[32] Individuals with an epiphyseal osteochondroma have a normal life expectancy. No malignant changes have been reported.
Enchondromatosis
Enchondromatosis can result in severe bone deformities, such as bowing and shortening. The risk of malignant change is high and is an indication for further intervention.
The prognosis of patients with enchondromatosis is difficult to assess. Most often, enchondromas are of no consequence, and patients are asymptomatic. The lesions are not life threatening and do not alter the person’s life expectancy. The two main factors that affect mortality and morbidity are painful malignant transformation and pathologic fracture, respectively. The prognosis may be better for patients with the dispersed forms of the disease than for those with localized forms, which may induce limb shortening or deformity.
Nonossifying fibroma
Most nonossifying fibromas regress, and no surgical intervention is required. Surgery is indicated for symptomatic lesions, as when patients present after sustaining pathologic fractures. Symptomatic lesions should first be treated conservatively. Conservative care consists of activity limitation and immobilization, in addition to yearly or biyearly radiography. If symptoms persist with conservative treatment, wide surgical curettage with autogenous bone graft is indicated.[61]
Nonossifying fibroma does not increase morbidity or mortality. For large, nonsymptomatic lesions, the only management indicated is yearly or biyearly radiographic examination until the lesion ceases growth and appears stable.
Special attention should be given to lesions larger than 33 mm and to those involving more than 50% of the diameter of the bone.[62] Attention is especially needed for lesions found in young children because of the risk of further growth.
Arata et al reviewed pathologic fractures in nonossifying fibromas over 49 years at the Mayo clinic.[62] Treatment consisted of cast immobilization, along with biopsy at a later date, simple curettage, curettage and autogenous bone-grafting, or segmental resection of fibular lesions.
Fibrous dysplasia
In patients with fibrous dysplasia, surgery is indicated as prophylaxis or treatment of pathologic fractures. With monostotic lesions, segmental resection can be performed.
Fibrous dysplasias progress in childhood but stabilize when skeletal maturity is achieved. Sarcomatous change occurs in 1.5% of patients.[63]
Hemangioma of bone
With hemangiomas of bone, intervention is indicated when patients become symptomatic. The symptoms determine the treatment, which can include irradiation, laminectomy, embolization, spinal reconstruction after tumor resection, or a combination of these procedures.
Most lesions are asymptomatic, and clinically significant symptoms develop in only 1-2% of patients. Because of the rarity of this lesion, no information about prognosis is available.[64]
Skeletal hemangiomatosis and lymphangiomatosis
In patients with skeletal hemangiomatosis or lymphangiomatosis, the lesions can regress, and surgical intervention may not be required. When treatment is needed, however, it usually involves curettage, grafting, and irradiation. Surgery may be needed to treat pathologic fractures or deformity.
Because skeletal hemangiomatosis and lymphangiomatosis are rare, each patient must be evaluated and treated on an individual basis, with his or her own prognosis. The prognosis improves for individuals without visceral involvement.
Copyright © www.orthopaedics.win Bone Health All Rights Reserved